Patología reumática en la adolescencia: casos clínicos
Patología reumática en la adolescencia: casos clínicos
R.M. Alcobendas Rueda, M.D, PhD.(1), C. Millán Longo, M.D.(2).
(1)Facultativo Especialista en Pediatría. Médico Adjunto de la Unidad de Reumatología Pediátrica. Hospital Universitario La Paz. Madrid. (2)Facultativo Especialista en Pediatría. Médico Adjunto del Servicio de Pediatría y la Unidad de Reumatología Pediátrica. Hospital Universitario La Paz. Madrid
Fecha de recepción: 23-12-2023
Fecha de publicación: 31 de marzo 2024
Adolescere 2024; XII (1): 88-96
Resumen
|
Las enfermedades reumatológicas en la infancia son variadas y diversas. Además de la sintomatología del paciente, la edad y el sexo son datos de interés relevante en la historia clínica pues permiten orientar de manera correcta la sospecha diagnóstica inicial. Se presentan a continuación dos casos representativos de la patología reumatológica en la adolescencia.. Palabras clave: Lupus eritematoso sistémico; Artritis idiopática juvenil; HLA-B27. |
Abstract
|
Rheumatological diseases in childhood are both varied and diverse. In addition to the patient’s symptomatology, age and sex are relevant data in the clinical history as they allow the initial diagnostic suspicion to be correctly assessed. Two representative cases of rheumatological pathology in adolescence are presented. Key words: Systemic lupus erythematosus; Juvenile idiopathic arthritis; HLA-B27. |
CASO CLÍNICO 1
Descripción del caso clínico
Paciente mujer de 12 años que ingresa en planta de hospitalización por edemas y fiebre para estudio. Refiere inicio de exantema en región malar bilateral 6 meses antes de consultar en el servicio de Urgencias hospitalario, que se extendió posteriormente a brazos y escote. Fue diagnosticada inicialmente de rosácea, sin mejoría con los tratamientos tópicos convencionales. En las últimas 2 semanas comienza con cefalea intermitente, dolor abdominal de tipo cólico, astenia, disminución del apetito, edema facial progresivo y, en los últimos 6 días, presenta febrícula diaria de máximo 37,5ºC axilar.
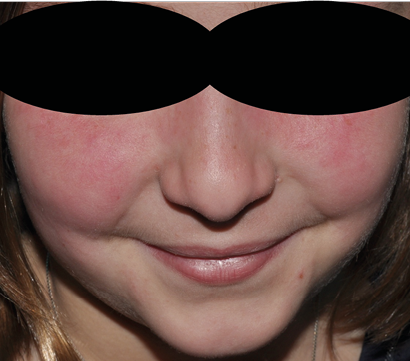
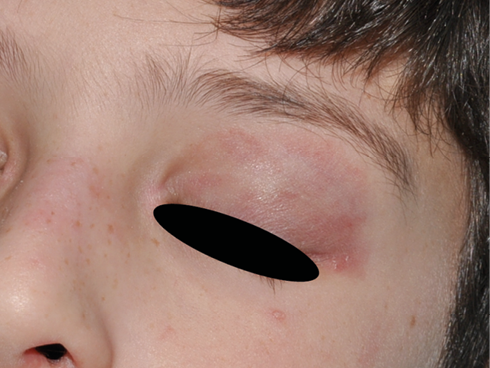
A su llegada a Urgencias está afebril y sus constantes vitales son las siguientes: FC 113 lpm, TA 109/68 mmHg, FR 26 rpm, SaO2 92 %. A la exploración presenta edema palpebral bilateral significativo (Figura 1), exantema en puente nasal y eritema malar bilateral, exantema micropapular eritematoso en escote e hiperemia en ambas palmas. Presenta disnea leve y a la auscultación pulmonar se evidencia hipoventilación en hemitórax izquierdo hasta campo medio. No se palpan visceromegalias ni adenopatías patológicas, no presenta signos de artritis y la paciente se encuentra neurológicamente asintomática, consciente y orientada y sin signos de focalidad neurológica.
Dados los signos y síntomas de la paciente, se canaliza una vía venosa periférica y se inicia oxigenoterapia en cánulas nasales convencionales a 2 L/min, además de extraer muestra para análisis de sangre. El análisis de sangre muestra los siguientes resultados: Hb 12,6 g/dL, leucocitos 4080/µcL, neutrófilos 2820/µcL, linfocitos 1060/µcL, plaquetas 138.000/µcL, coagulación normal, creatinina 0,53 mg/dL, transaminasas e iones normales, CPK 33 UI/L, albúmina 3,1 g/dL, PCR <0,5 mg/L. Se realiza una radiografía de tórax en la que se observa un derrame pleural izquierdo, que asciende a 4 cm en la ecografía pulmonar realizada, además de presentar discreta ascitis con cúmulos de líquido en localización subhepática y subesplénica en la ecografía abdominal.
Ante la descrita presentación clínica, y dada la sospecha diagnóstica de lupus eritematoso sistémico pediátrico (LESp), se decide ingreso para completar estudio e iniciar tratamiento.
Al ingreso en planta se completa el estudio con análisis de orina y estudio inmunológico, cuyos resultados se van conociendo en los días sucesivos: proteína/creatinina en orina de micción 446 mg/g creatinina (valor normal: <200 mg/g), IgG 1547 mg/dL (valor normal: 620-1150 mg/dL), IgA 184 mg/dL, IgM 137 mg/dL, C3 18,90 mg/dL (valor normal: >80 mg/dL), C4 0,6 mg/dL (valor normal: >15 mg/dL), CH-50 <50 UI/mL (valor normal: 150-9999 UI/mL), C1q no se detecta, anticuerpos ANA positivos 1/1280, anticoagulante lúpico, anticardiolipina y anti-β2-glicoproteína I negativos, anti-DNAds positivos >400 UI/mL (valor normal: <15 UI/mL), anti-histonas, anti-Ro y anti-La positivos y Coombs directo positivo.
La paciente es valorada por el servicio de Nefrología Pediátrica ante la sospecha de afectación renal por la enfermedad. Mantiene función renal y filtrado glomerular normales durante todo el ingreso, cifras de tensión arterial estables y mínima proteinuria con tendencia a negativizarse tras el inicio del tratamiento, sin hematuria ni otras alteraciones del sedimento urinario. Es valorada, asimismo, por el servicio de Cardiología Pediátrica. Se realizan electrocardiograma que resulta normal y ecocardiograma que no muestra alteraciones estructurales, derrame pericárdico ni anomalías de la función miocárdica. Se realiza además estudio oftalmológico que resulta anodino. Permanece en todo momento neurológicamente estable y asintomática.
Juicio clínico y diagnóstico diferencial
Con todo ello se diagnostica a la paciente de LESp. En la Tabla I se exponen los criterios de clasificación del LES, que no son específicos del LESp: los publicados en 2012 por la Systemic Lupus International Collaborating Clinics (SLICC) y los publicados en 2019 por la European League Against Rheumatism y la American College of Rheumatology (EULAR/ACR). Es importante resaltar que son criterios de clasificación y no de diagnóstico y, aunque deben servir de guía, el diagnóstico de la enfermedad no debe basarse únicamente en ellos.
A la hora de plantear el diagnóstico diferencial en un caso de LESp, es necesario descartar otras enfermedades que se puedan presentar con manifestaciones clínicas o alteraciones analíticas similares, como infecciones u otras enfermedades autoinmunes y conectivopatías.
Tratamiento y evolución
Ante la sospecha diagnóstica de LESp en la paciente, se inicia tratamiento con bolos de metilprednisolona intravenosa durante 3 días consecutivos y se continúa posteriormente con prednisona a dosis de 60 mg/día a partir del cuarto día. Dada la afectación de órgano con insuficiencia respiratoria hipoxémica con derrame pleural y que precisa oxigenoterapia suplementaria, a máximo 2,5 L/min, se inicia tratamiento de inducción inmunosupresor con ciclofosfamida intravenosa.
En contexto del tratamiento diurético con furosemida a dosis altas que recibe durante los primeros días, presenta tendencia a hipocalcemia e hipopotasemia, por lo que precisa suplementos de calcio y potasio y se sustituye la furosemida por espironolactona, que se suspende finalmente al octavo día de ingreso. Se inicia también tratamiento con hidroxicloroquina, además de suplementos de vitamina D y profilaxis antibiótica con cotrimoxazol.
Tras el tratamiento inmunosupresor inicial, presenta mejoría del estado general y de los edemas, la paciente queda afebril y se disminuye progresivamente la oxigenoterapia suplementaria coincidiendo con la mejoría respiratoria y del derrame pleural, hasta su retirada definitiva al séptimo día de ingreso. Dada la evolución favorable, se decide el alta hospitalaria con control posterior estrecho en consultas externas de Reumatología Pediátrica.
Discusión
En el LESp la mortalidad en ausencia de tratamiento es del 95 % a los 5 años
El caso clínico que se presenta constituye un ejemplo típico de presentación del LES en la edad pediátrica. El LESp es una enfermedad crónica que se caracteriza por la presencia de múltiples autoanticuerpos, afectación multisistémica y curso variable. La mortalidad estimada en ausencia de tratamiento del 95 % a los 5 años. En nuestro medio se estima una incidencia de 0,3-0,9 casos/100.000 niños/año y una prevalencia de 3,5 casos/100.000 niños. La edad media de debut son los 12 años y es más frecuente en mujeres y en raza no caucásica (afroamericanos, hispanos y asiáticos). Se debe sospechar LESp en un paciente con síntomas sugestivos y alteraciones analíticas compatibles, como se ha expuesto en apartados anteriores.
El objetivo principal del tratamiento es lograr la inactividad de la enfermedad y mantenerla en el tiempo. Además, evitar el daño y las complicaciones derivadas de los tratamientos empleados, reduciendo en lo posible la toxicidad farmacológica, es otro de los objetivos a largo plazo. El tratamiento se divide en una fase inicial de inducción y una siguiente fase de mantenimiento.
Los principales fármacos empleados son los siguientes:
- Glucocorticoides: de elección en la fase aguda. Se administran inicialmente en forma de bolos intravenosos (1-5 días) y posteriormente se reduce progresivamente la dosis de forma individualizada a medio-largo plazo.
- Hidroxicloroquina: terapia de mantenimiento en todos los casos. Es recomendable realizar revisiones oftalmológicas periódicas por el riesgo de alteraciones oculares que conlleva su uso de forma prolongada.
- Inmunosupresores: metotrexato (para síntomas cutáneos y articulares), azatioprina (tratamiento de mantenimiento), micofenolato de mofetilo (tratamiento de inducción y de mantenimiento, como alternativa a la azatioprina o de primera elección en manifestaciones graves), ciclofosfamida (tratamiento de inducción en manifestaciones graves o con riesgo vital como enfermedad renal, cardiopulmonar y neurológica). Durante el tratamiento con ciclofosfamida se debe valorar el tratamiento para protección ovárica por el riego de fallo ovárico prematuro en mujeres en edad fértil.
- Otros tratamientos: fármacos biológicos (rituximab, belimumab), que se emplean en función de la afectación clínica y de la gravedad; ácido acetilsalicílico (para tratamiento del síndrome antifosfolípido asociado al LESp).
Se exponen a continuación unas recomendaciones generales aplicables a todos los pacientes con LESp:
- Es deseable evitar la exposición solar directa y utilizar fotoprotectores de alto índice de protección.
- Se recomienda realizar ejercicio físico regular y adecuado a cada caso.
- Se recomienda evitar el consumo de sustancias tóxicas, como el tabaco, el alcohol y otras sustancias.
- La dieta debe ser sana, variada y equilibrada y es recomendable controlar el aporte calórico, sobre todo durante el tratamiento con glucocorticoides, y restringir la ingesta de grasas en caso de dislipemia. Se recomienda, además, la suplementación con calcio y vitamina D con el objetivo de prevenir la osteoporosis secundaria al tratamiento prolongado con glucocorticoides.
- Están contraindicadas las vacunas de virus vivos durante el tratamiento inmunosupresor. Además, se recomienda reforzar la pauta vacunal antineumocócica con la vacuna polisacárida 23-valente, así como administrar las vacunas frente a meningococo B y ACWY y la gripe estacional.
- Se debe recomendar el uso de métodos anticonceptivos adecuados para evitar embarazos no deseados durante el tratamiento. Es importante tener en cuenta que los anticonceptivos hormonales están desaconsejados en algunos casos por su riesgo protrombótico asociado.
Los anticonceptivos hormonales están desaconsejados en algunos casos de LESp por su riesgo protrombótico
CASO CLÍNICO 2
Paciente varón de 12 años acude a consulta derivado por su pediatra de atención primaria porque hace 3 meses comenzó con dolor en región lumbar baja derecha irradiado a nalga de características mixtas, siendo peor por la mañana con cojera y dificultad en las primeras horas del día con mejoría posterior, pero también limitando su participación en los partidos de fútbol por aparición del dolor. Como pruebas complementarias aporta radiografía lumbar y anteroposterior de pelvis sin alteraciones y una RMN lumbosacra solicitada por traumatología en la que se evidencia una sacroilitis derecha compatible con etiología inflamatoria. Ha recibido tratamiento con ibuprofeno, con discreta mejoría pero sin resolución del cuadro. Asimismo, en las últimas semanas se ha asociado tumefacción y dolor en tobillo izquierdo que ha ido empeorando de manera progresiva. Afebril, no antecedentes traumáticos, no ojo rojo ni alteración de consistencia o características de las heces. Como único antecedente de interés, presenta un abuelo por rama materna con Espondilitis anquilosante HLA B27 positivo sin tratamiento en la actualidad. No antecedentes famililares de psoriasis ni de enfermedad inflamatoria intestinal.
En la exploración física se objetiva tumefacción, limitación y dolor en tobillo y tarso izquierdo, así como maniobras sacroilíacas positivas en lado derecho, siendo el resto de la exploración articular y por aparatos normal.
Ante la sospecha de debut de Artritis idiopática juvenil (artritis crónica de >6 semanas de evolución en <18 años) se completa el estudio con las siguientes pruebas complementarias:
- Analítica de sangre: Leucocitos 7690/mcL (4800-15000), Hb 12,8 g/dl (10,6-14,6), vcm 83,5 fL (72-93), plaquetas 332 000/mcL (180 000- 490 000), neutrófilos 4740/mL (1500-8700), linfocitos (2700-9000), VSG 30 mm/h, GOT 17 UI/L (<95), GPT 15 UI/L (<35), PCR 7,5 mg/dl (0-0,5), creatinina 0,44 mg/dl (0,3-0,7), hierro 43 mcg/dl (50-120), IST 16 % (15-50).
- Inmunología: ANA, FR, ASCA y ANCA negativo. HLA B27 positivo.
- Mantoux: negativo.
- Serologías: Rubeola IgG: positivo. Sarampión IgG: positivo. Parotiditis IgG: positivo. Varicela Ig G: positivo. VHA IgM: negativo. HBs Ag: negativo. Anti-HB Core: negativo. Anti HBs Ag: negativo. VHC: negativo. VIH 1/2 AG-AC: negativo.
- Calprotectina fecal: 45 mcg/g.
- Ecografía de tobillo y pie derecho: compatible con artritis de tobillo y tarso.
Juicio clínico
De acuerdo a los últimos criterios de AIJ (nueva clasificación de la AIJ, consenso internacional de PRINTO 2019) (Tabla II) es catalogado como Entesitis/espondilitis relacionada con AIJ (tradicionalmente conocida como Artritis relacionada con entesitis) HLA B27 positivo, ANA, FR negativo.
Tratamiento y evolución
Debido a la persistencia de la sintomatología y a la afectación axial, tras realizar despistaje de tuberculosis se inicia tratamiento con adalimumab 40 mg/quincenal subcutáneo, metotrexato en comprimidos (15 mg/semana) y ácido fólico (5mg/semana dos días después de la administración del metotrexato). Asimismo, se realiza infiltración de tobillo izquierdo bajo sedación, con triamcinolona 40 mg.
Además, se le explican pautas de vacunación especialmente indicadas en este tipo de pacientes y tratamiento, recomendando especialmente la vacunación en época epidémica de gripe y COVID-19, así como la revacunación con dosis extra de VHB puesto que el estudio de respuesta postvacunal, aunque no es cuantitativo, es negativo (Anti HBs Ag: negativo).
Se indica qué actitud tomar ante episodio febril, omitiendo la dosis de tratamiento (adalimumab o metotrexato) si el día de la administración presenta fiebre, se advierte evitar hábitos tóxicos y se informa sobre la potencial teratogenicidad del metotrexato.
En consultas sucesivas presenta mejoría progresiva de la sintomatología y analítica (normalización del hierro) hasta su completa normalización con incorporación total a la actividad física deportiva, por lo que el tratamiento se mantiene.
Discusión
La Entesitis/espondilitis relacionada con AIJ (tradicionalmente conocida como Artritis relacionada con entesitis) se engloba junto con la Artritis psoriásica, las Artritis reactivas y las Artritis asociadas a Enfermedad inflamatoria intestinal (EII) y las Artritis indiferenciadas como posibles formas de debut de Espondiloartropatía en la infancia. Como grupo, las Espondiloartropatías, se caracterizan por presentar una serie de manifestaciones clínicas particulares que las diferencian del resto como son el compromiso axial, artritis periférica, entesitis, tarsitis, dactilitis, manifestaciones extraarticulares (uveítis sintomáticas, psoriasis, EII) y probable asociación con el antígeno HLA B27. Aunque generalmente debutan en la edad adulta (20-40 años), es necesario conocer que hasta un 10-20 % comienzan en la infancia.
La Entesitis/espondilitis relacionada con AIJ supone un 15-20 % del total de pacientes diagnosticados con AIJ. Esta categoría se presenta más frecuentemente en varones mayores de 6 años y suele asociar la presencia del antígeno HLA-B27. A diferencia del adulto donde es típico el dolor lumbar con afectación de la columna, en el niño la forma de presentación suele ser en forma de artritis periférica de grandes articulaciones y asimétrica, siendo muy sugerente la artritis de cadera en paciente mayor. En caso de presentación axial, la afectación de sacroilíacas uni o bilateral es la norma y suele aparecer en niños pre o adolescentes.
Las molestias asociadas a la entesitis (al igual que la sacroilitis) pueden tener también ritmo mecánico por sobreuso/sobrecarga y es típico que empeoren con el ejercicio
Asimismo, las entesitis típicas del paciente pediátrico suelen ser la inserción del tendón rotuliano a nivel de la tuberosidad anterior (entesitis rotuliana), entesitis del tendón de Aquiles a nivel del calcáneo (entesitis aquílea) y fascitis plantar. La entesitis se manifiesta en la exploración como punto doloroso a la presión sobre el sitio de inserción de los tendones, ligamentos o fascias a nivel óseo y puede asociar a nivel rotuliano o aquíleo tumefacción e incluso eritema en fases de mayor inflamación. Aunque el ritmo de dolor en los pacientes con AIJ es característicamente inflamatorio, es necesario conocer que las molestias asociadas a las entesitis (al igual que las sacroilitis) pueden tener también ritmo mecánico por sobreuso/sobrecarga y es típico que empeoren con el ejercicio y lleguen a limitar sus actividades deportivas. Este hecho hace que en ocasiones exista un retraso diagnóstico importante, siendo erróneamente catalogados como tendinitis.
La afectación de tarso puede ser mixta asociando artritis del medio y región proximal del antepie, entesitis y tenosinovitis. En la exploración física se manifiesta como tumefacción y en ocasiones eritema, pero sobre todo con dolor y retirada a la presión directa del tarso. Su afectación es típica y de difícil abordaje, por lo que en ocasiones requieren asociar tratamiento con corticoide oral y en caso de no mejorar con FAME, estaría indicada la introducción de terapia biológica.
Un pequeño porcentaje de pacientes, a diferencia del resto de pacientes con AIJ (excluyendo la AIJ sistémica o AIJ con importante afectación poliarticular), pueden asociar fiebre-febrícula y elevación de reactantes de fase aguda al debut, en los que sería necesario realizar un correcto diagnóstico diferencial.
Asimismo, otra particularidad es que las uveítis asociadas a esta categoría de AIJ suelen ser sintomáticas en forma de ojo rojo doloroso, aunque igualmente se recomiendan valoraciones periódicas por oftalmología.
Como forma de espondiloartropatía juvenil, es importante descartar la asociación personal o familiar con psoriasis y enfermedad inflamatoria intestinal. Se recomienda añadir al cribado ASCA y calprotectina fecal incluso estando asintomáticos, puesto que puede tratarse de una forma de debut de EII, así como estar pendientes durante su evolución de manifestaciones digestivas (improbable a su vez si se encuentran con tratamiento biológico con adalimumab e infliximab pero que podrían aparecer en el espaciado o retirada de la medicación).
El antecedente de diarrea en caso de presentarse, máxime si asocia productos patológicos, obliga a descartar artritis reactiva descartando infección gastrointestinal (característicamente por Yersinia, Salmonella y Campylobacter). Además, y sobre todo por el aumento de relaciones sexuales en la infancia, se debería interrogar sobre uretritis/balanitis y en caso de sospecha añadir serología de Chlamydia trachomatis y Ureaplasma urealyticum. Actualmente no está recomendado añadir el despistaje al debut de paciente con AIJ, el estudio de Gonococco. Sin embargo, debido a los cambios de prácticas en el momento actual, posiblemente haya que replantearse en los próximos años añadir su estudio al despistaje inicial de artritis en pre-adolescentes, al igual que se realiza de manera rutinaria en pacientes adultos.
El tratamiento biológico con Anti-TNF debería considerarse como terapia inicial en pacientes que presenten afectación en articulaciones de riesgo (columna cervical, muñeca, cadera) o sacroilitis o entesitis, así como alternativa al tratamiento con FAME cuando no hay respuesta a estos o una elevada actividad inflamatoria
En el paciente del caso clínico se decide la introducción conjunta de FAME con fármaco biológico puesto que ningún FAME ha resultado ser eficaz en la afectación axial. Asimismo, el tratamiento biológico con Anti TNF debería ser considerado como terapia inicial en pacientes que presenten afectación de articulaciones de riesgo (columna cervical, muñeca, cadera) o sacroilitis o entesitis, así como alternativa al tratamiento con FAME cuando no hay respuesta a estos o cuando hay una elevada actividad inflamatoria.
Tablas y figuras
Tabla I. Criterios de clasificación de Lupus Eritematoso Sistémico
|
Criterios SLICC 2012 |
EULAR/ACR 2019 |
|
4 criterios: ≥1 clínico + ≥1 inmunológico o Nefritis lúpica en biopsia + ANA o |
ANA (+) ≥1/80 + ≥10 puntos (incluyendo al menos un criterio clínico) *Dentro de cada dominio, únicamente se tiene en cuenta el criterio de puntuación más alta. |
|
A. CRITERIOS CLÍNICOS
B. CRITERIOS INMUNOLÓGICOS
|
1. DOMINIOS CLÍNICOS Y CRITERIOS Constitucional Fiebre >38,3°C 2 Hematológico Leucopenia <4,0 x 109/L 3 Trombopenia <100 x 109/L 4 Hemólisis autoinmune 4 Neuropsiquiátrico Delirio 2 Psicosis 3 Crisis epilépticas 5 Mucocutáneo Alopecia no cicatricial 2 Úlceras orales 2 Lupus subcutáneo o discoide 4 Lupus agudo 6 Serositis Derrame pleural o pericárdico 5 Pericarditis aguda (≥2 episodios) 6 Musculoesquelético Sinovitis ≥2 articulaciones o dolor ≥2 articulaciones y rigidez matutina de >30 minutos 6 Renal Proteinuria ≥0,5 g/24h 4 Biopsia: nefritis lúpica clase II o V 8 Biopsia: nefritis lúpica clase III o IV 10 2. DOMINIOS INMUNOLÓGICOS Y CRITERIOS Anticuerpos antifosfolípidos positivos Anticardiolipina, anti-β2- glicoproteína I o anticoagulante lúpico 2 Complemento Diminución C3 o C4 3 Disminución C3 y C4 4 Anticuerpos específicos para LES Anti-DNAds o anti-Sm 6 |
|
ACR: American College of Rheumatology; DNAds: DNA de doble cadena; EULAR: European League Against Rheumatism; SLICC: Systemic Lupus International Collaborating Clinics. |
|
Tabla II. Criterios de entesitis/espondilitis relacionada con la AIJ
|
Nueva clasificación de la AIJ, Consenso Internacional de PRINTO 2019.
Figura 1. Caso clínico 1: edema facial y eritema malar y en puente nasal
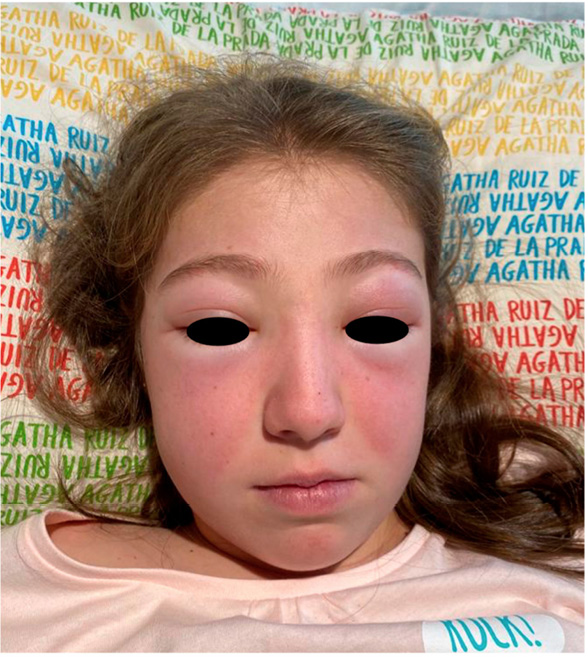
Elaboración propia.
Bibliografía
- Klein-Gitelman M, Lane JC. Systemic Lupus Erythematosus. En: Petty RE, Laxer RM, Lindsley CB, Wedderburn LR, eds. Textbook of pediatric rheumatology. 7th edition. Philadelphia: Elsevier; 2016; p.285-317.
- Boteanu A. Lupus eritematoso sistémico pediátrico. Protoc diagn ter pediatr. 2020;2:115-128.
- Urbaneja Rodríguez E. Lupus y otras conectivopatías en la infancia. Pediatr Integral 2022;XXVI(3):163-174.
- García MI, Camacho M. Artritis relacionada con entesitis. Artritis psoriásica. Protoc diagn ter pediatr. 2020;2:77-88.
- Martini A, Ravelli A, Avcin T, Beresford MW, Burgos-Vargas R. Cuttica R, et al. Toward new classification criteria for juvenile idiopathic arthritis: first steps, Pediatric Rheumatology International Trials Organization International Consensus. J Rheumatol. 2019;46:190-7.
No existen conflictos de interés en la realización de este artículo.