Sesión II Actualización: Obesidad en el adolescente. Caso clínico: Obesidad y talla baja
Sesión II Actualización: Obesidad en el adolescente
Caso clínico: Obesidad y talla baja
M. Güemes Hidalgo.
Pediatra. Doctora en Medicina. Servicio de Endocrinología. Hospital Infantil Universitario Niño Jesús. Madrid.
Adolescere 2023; XI (2): 41-46
Resumen
|
Se presenta el caso de una adolescente con obesidad y estancamiento de la talla desde hace varios años, siendo diagnosticada, a raíz de pérdida brusca de la visión, de tumor del sistema nervioso central y deficiencia hormonal múltiple. Se presentan los datos diagnósticos, la evolución y el tratamiento. Palabras clave: Obesidad; Hipocrecimiento; Germinoma. |
Abstract
|
The case of an adolescent female with obesity and stature stagnation for several years is presented. She was diagnosed with a central nervous system tumor and multiple hormone deficiency after a sudden loss of vision. Diagnostic data, progression and treatment are presented. Key words: Obesity; Failure to thrive; Germinoma. |
Anamnesis
Adolescente controlada desde los 11 años por endocrinólogo infantil en centro privado por obesidad y estancamiento de la talla. Respecto al peso, refieren que éste había gradualmente aumentado desde los 5 años aproximadamente, manteniéndose en percentil 75 hasta los 11 años, y ascendiendo en los últimos 2 años al percentil 90. Comentaban los padres que la joven siempre había seguido una dieta mediterránea cuidada, comida casera, con ingesta de refrescos/aperitivo/comida rápida “excepcionalmente”, y realizando como deporte la educación física escolar junto con 2h a la semana de tenis. Según los padres, en los últimos años el apetito se había vuelto mucho mayor, con menor sensación de saciedad.
Aportan tallas hasta los 6,5 años en percentil 97, disminuyendo desde entonces hasta el percentil 25-50 a los 11 años, y posteriormente al percentil 10 a los 13 años 7 meses. Refieren “analítica hormonal” previa sin alteraciones.
La Figura 1 muestra los datos antropométricos disponibles.
Acude a Urgencias de nuestro hospital a los 13 años 7 meses por pérdida de visión progresiva de 3 semanas, tanto de cerca como de lejos (constatada en examen por Oftalmólogo). Así mismo, presenta poliuria y polidipsia, indicando que bebe entre 4-6 litros de agua diarios, y que cada noche se levanta 4-5 veces para orinar y beber. Refiere cansancio, pero no cefalea, ni vómitos, ni otras alteraciones neurológicas. (Figura 1: Datos disponibles de talla y peso de la paciente, empleando curvas de Hernández y colaboradores del año 1988). La talla genética o diana corresponde a 174 +/-5cm.
Antecedentes
- Antecedentes familiares: Madre: española, sana, talla: 173 cm, peso: 70kg, GAV: 2-0-2, menarquia: 12,5 años. Padre: español, sano, talla: 188 cm, peso: 93kg, desarrollo puberal
tardío. Sin consanguinidad. Hermano: 16 años, 188 cm, “normopeso”, madurador precoz, sano.
Sin otros antecedentes familiares de interés. - Antecedentes personales: Embarazo controlado, sin alteraciones. Parto eutócico en semana 36+1. PRN: 2700g (p63, 0.34 DE). Longitud: 47 cm (p54, 0.11 DE). Periodo neonatal sin incidencias. Cribado endocrino – metabólico normal. Sin enfermedades previas, salvo valoración hasta los 13 años 2 meses por endocrinología por detectar desde los 11 años obesidad y estancamiento de talla.
Exploración física
Edad: 13 años 7 meses. Peso: 55 kg (p88, 1.18 DE). Talla: 144 cm (p7, -1.55 DE). IMC: 26.52 % (p98, 2.11 DE). Talla genética: 174 ±5 cm. Tensión arterial: Sistólica: 105 mm Hg (p54, 0.11 DE). Diastólica: 65 mm Hg (p59, 0.24 DE). Buen estado general, bien hidratada y perfundida, eupneica. Hábito pícnico, sin aspecto cushingoide, ni estrías. Fenotipo normal, armónico. En el examen neurológico todo normal, salvo: amaurosis en ojo izquierdo (refiere que distingue figuras, aunque borrosas). Dificultad para distinguir letras a 20 cm. Campimetría por confrontación alterada con dificultad/incapacidad para visualizar objetos en campo fronto-temporal derecho y temporal izquierdo (mayor afectación de lado derecho). Auscultación pulmonar: buena entrada de aire bilateral. Auscultación cardiaca: rítmica, sin soplos. Abdomen: blando, depresible, sin masas ni megalias. ORL: sin alteraciones. Genitales externos femeninos normales en estadio puberal I de Tanner (Telarquia1, Pubarquia2, Axilarquia1).
Ante la clínica referida se solicitan las siguientes pruebas complementarias:
- TC cráneo: En la región supraselar se identifica una gran masa de aspecto sólido y bordes bien definidos, que se extiende hacia ambos lóbulos frontales, aunque de predominio hacia el lado izquierdo, donde sus límites son más imprecisos, rodeando a las astas frontales de los ventrículos laterales. La lesión parece englobar al quiasma y al tallo hipofisario y podría extenderse hacia el interior de la silla turca, aunque de forma discreta. Sistema ventricular de morfología y tamaño normal. No hay signos de sangrado intra ni extraaxial.
- RM cráneo espinal: Se visualiza una lesión centrada en región supraselar que engloba el quiasma óptico, la porción prequiasmática de ambos nervios ópticos y el tallo hipofisario.
La lesión es sólido-quística; las medidas aproximadas son de 24 x 24 mm la porción supraselar (APxT) y aproximadamente 48 mm de eje craneocaudal. La línea media está centrada. No se aprecia ventriculomegalia. No muestra signos de diseminación a distancia. - Analítica de sangre (con Dexametasona):
T4L ** 0.48 ng/dl (0.65 – 1.4), TSH 1.79 UI/ml (0.36 – 5.5), LH ** 0.01 mUI/ml (0.2 – 15), FSH ** 0.07 mUI/ml (2 – 22), Prolactina 13.1 ng/ml (3.5 – 25), Estradiol * 8.2 pg/ml (10 – 400), Cortisol ** 0.4 g/dl (5.9 – 22), ACTH * 3.1 pg/mL (6 – 48), IGF I * 76 ng/ml (105 – 516), IGFBP-3 2.54 g/ml (2.32 – 6.19). Glucosa 110mg/dl (<100), Insulina ** 25.5 IU/ml (4 – 11), Ácido Úrico * ٧.٨٥ mg/dL (٢.٧ – ٥.٦), Colesterol total 129 mg/dL (120 – 200), Colesterol HDL * 25 mg/dL (40 – 65), Colesterol LDL 60.4 mg/dL (60 – 130) Colesterol VLDL * 43.6 mg/dL (6.5 – 28), Trigliceridos * 218 mg/dL (35 – 135). Alfafetoproteína 2,19 ng/ml (1-15), B-HCG total <1.2 mIU/mL (0.01 – 5).
- En L.C.R.: Beta-HCG total ** 21.6 mUI/mL (0.01 – 5).
- En el contexto de restricción hídrica incidental, presenta en analítica sanguínea sodio de 157 mEq/l, poliuria (7ml/kg/h) y marcada avidez por el agua. Lengua pastosa en la exploración física. Tras la administración de desmopresina 60mcg vo: sodio plasmático de 153 mEq/l, diuresis 5cc/kg/h.
- Edad ósea (método Greulich & Pyle): 11 años – Edad cronológica 13 años 10 meses.
- Test de LH-RH:
- FSH (mlU/ml): 0.19 (-30 min) – 0.29 (basal) – 1.38 (30min) – 1.39 (60min) – 1.56 (90min) – 2.34 (120 min).
- LH (mlU/ml): 0.03 (-30 min) – 0.05 (basal) – 0.09 (30min) – 0.11 (60min) – 0.19 (90min) – 0.21 (120 min).
- Densitometría: L1-L4 Z-score -3.8 DS
Evolución clínica
La paciente ingresa desde Urgencias con dexametasona intravenosa (para disminuir el edema cerebral peritumoral).
Durante su ingreso se confirma deficiencia de la hormona antidiurética hipofisaria (diabetes insípida central) ante los datos clínicos de poliuria y polidipsia, con hipernatremia al restringir la ingesta de agua libre, junto con el descenso de la natremia y de la poliuria al administrar desmopresina sublingual. Las concentraciones bajas de T4L junto con TSH inapropiadamente normal son compatibles con hipotiroidismo central, que explica el cansancio experimentado por la paciente y la aparición de la obesidad desde dos años antes.
La velocidad de crecimiento subóptima, “estancamiento de talla”, junto con concentraciones bajas de factores de crecimiento, IGF-1 (insulin-like growth factor 1) e IGFBP-3 (insulin-like growth factor binding protein 3), apuntan a una deficiencia de hormona de crecimiento. Al tener la paciente una edad cronológica superior a los 13 años, con una edad ósea de 11 años (suele coincidir con inicio de la pubertad) sin inicio puberal y con gonadotropinas basales casi indetectables, sugiere hipogonadismo central, confirmado posteriormente con el test de estímulo de LHRH.
En este contexto es probable que la paciente además presente un déficit de ACTH, pero éste último está pendiente de confirmar bioquímicamente, tras la retirada completa de corticoterapia exógena. Se realiza ventriculoscopia con toma de biopsia, mostrando la anatomía patológica una neoplasia maligna germinal de tipo germinoma.
Diagnósticos
- Germinoma del sistema nervioso central
- Deficiencia hipofisaria múltiple: diabetes insípida central, hipotiroidismo central, hipogonadismo central, deficiencia de hormona de crecimiento y probable déficit de ACTH
- Obesidad. Comorbilidades: dislipemia, hiperinsulinemia, hiperuricemia
- Estancamiento de la talla
- Densidad mineral ósea disminuída. Hiperpatiroidismo secundario a hipovitaminosis D
Diagnóstico diferencial
Los astrocitomas, tumores derivados de las células tipo astrocito, son los tumores del sistema nervioso central, más frecuentes en la edad pediátrica
Dado el hallazgo en la RMN craneal, entre los diagnósticos diferenciales se deben considerar, principalmente, los siguientes tumores del sistema nervioso central: glioma quiasmático/hipotalámico, tumor germinal del tallo hipofisario, craneofaringioma, astrocitoma hipotálamo-quiasmático.
Los astrocitomas, tumores derivados de las células tipo astrocito, son los tumores del sistema nervioso central, más frecuentes en la edad pediátrica, representando hasta el 50 % de los casos. Los craneofaringiomas suponen un 5.6 % y los tumores de células germinales un 2.5 %.
Tratamiento
Como terapia oncológica, recibió 4 ciclos de quimioterapia según el esquema ACNS 1123 (incluye carboplatino y etopósido), seguido de protonterapia holocraneal (dosis: 24 Gy). La protonterapia es un tipo de radioterapia que aplica protones, permitiendo aumentar la precisión y mayor efecto antitumoral sobre el tejido a radiar y produciendo un menor daño de los tejidos peritumorales sanos. Tras lo cual, por pruebas de imagen y marcadores tumorales, es considerada en remisión completa hasta la actualidad (12 meses). Como tratamiento hormonal sustitutivo recibe desde su ingreso, en cuanto se realizaron los diagnósticos de diabetes insípida e hipotiroidismo central, desmopresina sublingual con acceso libre al agua y levotiroxina oral. Ante hipogonadismo
hipogonadotropo, a los 14 años se comenzó la inducción puberal con dosis ascendentes de 17-beta-estradiol en forma de parches cutáneos. Dado que lleva 1 año en remisión completa, con buen pronóstico de talla, se decidió, por el momento, no sustituir la deficiencia de GH. Podrá comprobarse la integridad/afectación del eje hipotálamo-hipófiso-suprarrenal cuando se haya retirado la corticoterapia completamente, mediante un test de ACTH o hipoglucemia insulínica. Debido a presentar una densitometría que muestra densidad mineral ósea baja, junto con valores de vitamina D 13,5 ng/ml (20 – 60) y PTH 142 pg/ml (10 – 55), recibe tratamiento con vitamina D oral.
Discusión del caso
La etiología del sobrepeso/obesidad tradicionalmente se ha dividido en causas exógenas (representan más del 90 % de los casos) y endógenas (menos del 10 % de casos, incluyendo obesidades monogénicas, síndromes polimalformativos y endocrinopatías)
Se presenta el caso de una adolescente con obesidad y franco estancamiento de talla de varios años de evolución. En nuestro medio, se tiende a definir sobrepeso si el índice de masa corporal (IMC: kg/cm2) se encuentra entre los percentiles 90-97, y obesidad si supera el percentil 97, según edad y sexo referido a los datos y curvas de Hernández y colaboradores del año 1988. La etiología del sobrepeso/obesidad tradicionalmente se ha dividido en causas exógenas (representan más del 90 % de los casos, debidos a la combinación de: incremento del aporte energético, disminución del consumo energético e influencia poligénica) y endógenas (menos del 10 % de casos, incluyendo obesidades monogénicas, síndromes polimalformativos y endocrinopatías). En la paciente actual el ascenso ponderal no había sido brusco (para sospechar, por ejemplo, un hipercortisolismo endógeno), sino mantenido en el tiempo, de inicio en la infancia (no en los primeros años de vida, como para sospechar causa monogénica), sin estigmas fenotípicos (para sospechar un síndrome polimalformativo), habiéndose atribuido a la obesidad más frecuente hoy en día en nuestras consultas, la multifactorial por desequilibrio energético. La aproximación diagnóstica a la obesidad de presentación tras los 5 años, de carácter progresivo -no brusco-, incluye, además de los antecedentes familiares y personales, una minuciosa encuesta de los hábitos alimentarios y de la actividad física, además de búsqueda de posibles comorbilidades [metabólicas (metabolismo de los carbohidratos, lípidos, hiperuricemia), cardiovasculares (hipertensión arterial, arteriosclerosis), ortopédicas (ej: enfermedad de Blount, enfermedad de Legg-Calvé-Perthes), emocionales (imagen corporal, depresión,…), miscelánea (pseudotumor cerebrii, pancreatitis, intertrigo, glomerulopatía,…)].
En el caso concreto de la paciente, un dato clave asociado a la obesidad, que cambia el enfoque etiológico radicalmente, es la asociación con una disminución de la velocidad de crecimiento o estancamiento de la talla.
Es criterio de hipocrecimiento una velocidad de crecimiento inferior a -1 desviación estándar para la edad y sexo, mantenida durante al menos 2 años, independiente de la talla actual
Una velocidad de crecimiento inferior a -1 desviación estándar (DE) (<p25) para su edad y sexo, mantenida durante al menos 2 años, independientemente de la talla actual, es criterio de hipocrecimiento. Si bien, la paciente de nuestro caso no es una talla baja sensu estricto (dicha definición exige estatura <-2 DE o inferior al percentil 3 para edad y sexo de su población de referencia), sí que en su presentación en la Urgencia tenía una talla baja para su talla genética, y un llamativo parón de crecimiento (Figura 1), que exige estudio etiológico. De cara al diagnóstico, además de los antecedentes familiares (otros familiares con talla baja, endocrinopatías,…), personales (antropometría neonatal, patrón nutricional, cuándo inició la talla baja…), la exploración física será determinante para clasificar la posible etiología del hipocrecimiento en proporcionado o no (estigmas fenotípicos, medir segmentos, bocio, aspecto cushingoide,…).
Las pruebas diagnósticas de primer nivel incluyen: edad ósea y analítica con hemograma, velocidad de sedimentación globular, función hepato-renal, cribaje de celiaquía, función tiroidea y factores de crecimiento, IGF-1 e IGFBP-3. En el caso de nuestra paciente, la velocidad de crecimiento patológica, con factores de crecimiento bajos, apunta a una deficiencia de hormona de crecimiento. La confirmación bioquímica de este déficit requiere de test/s de estímulo de GH, entre los que se encuentran: clonidina, glucagón, hipoglucemia insulínica, entre otros. Todo déficit de GH precisa de prueba de imagen cerebral, siendo la más empleada para caracterización la RMN cerebral centrada en región hipotálamo-hipofisaria.
En el caso actual, lamentablemente, aunque había sido valorada por un especialista, no es hasta 2 años después que, por pérdida aguda de visión, es diagnosticada de un tumor del sistema nervioso central. Entonces, se realiza el diagnóstico de múltiples deficiencias hormonales: diabetes insípida, hipotiroidismo, hipogonadismo, déficit de GH y probable déficit de ACTH. La paciente recibe tratamiento sustitutivo de estas deficiencias, a excepción de GH. El motivo de no sustituir GH es porque sólo ha pasado un año desde que la paciente está en remisión completa y que presenta una buena expectativa de talla final (actualmente tiene una talla de 157 cm con una edad ósea de 13 años 3 meses y todavía tiene por delante evolución de la inducción puberal, durante la cual está creciendo por influencia del efecto de las hormonas sexuales promoviendo el crecimiento).
La obesidad más frecuentemente encontrada en la práctica clínica es multifactorial, pero no se debe olvidar que existen causas orgánicas de la misma, siendo algunas de ellas muy graves
El presente caso ilustra muchos aspectos de los que aprender. En primer lugar, la relevancia de realizar una minuciosa historia clínica, para intentar un diagnóstico precoz y minimizar secuelas; destaca poliuria y polidipsia desapercibidas durante tres años. Aunque la obesidad más frecuentemente encontrada en la práctica clínica es multifactorial, no se debe olvidar que existen causas orgánicas de la misma, siendo algunas de ellas muy graves. No está claro en qué momento se manifestó el hipotiroidismo, pues parece lógico pensar que haya contribuido, al menos en parte, al ascenso ponderal. El “mayor apetito” referido por los padres en los últimos años, posiblemente es reflejo de afectación del tumor sobre el centro hipotalámico de regulación del apetito, condicionando su obesidad. El déficit de GH probablemente también participe en su obesidad, al poseer la hormona de crecimiento efecto anabólico y lipolítico. Así mismo, con los datos retrospectivos de tallas previas, se visualiza un patrón muy sugestivo de deficiencia de hormona de crecimiento. También, destacar la necesidad de estudio de una adolescente que no ha iniciado caracteres sexuales por encima de la edad de 13 años. Se resalta la importancia de no impedir el acceso al agua libre ante sospecha/confirmación de diabetes insípida central con mecanismo de la sed preservado. La administración de corticoides a dosis elevadas suprime el eje hipotálamo-hipófiso-gonadal, impidiendo en ese momento el estudio de idoneidad/deficiencia de ACTH. El hipercortisolismo produce ascenso ponderal marcado, y su combinación con hipoestrogenismo, deficiencia de GH, inmovilidad e hipovitaminosis D, contribuyen al descenso de densidad mineral ósea.
Tablas y figuras
Figura 1. Datos disponibles de talla y peso de la paciente, empleando curvas de Hernández y colaboradores del año 1988
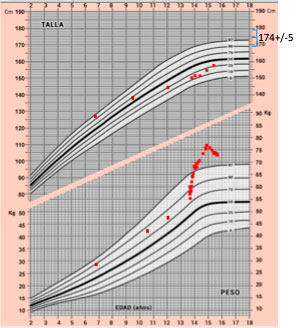
La talla genética o diana corresponde a 174 +/-5cm.
Bibliografía
- Partenope C, Pozzobon G, Weber G, Arya VB, Carceller F, Albanese A. Endocrine manifestations of paediatric intracranial germ cell tumours: from diagnosis to long-term follow-up. Endocrine. 2022 Sep;77(3):546-555. doi: 10.1007/s12020-022-03121-9. Epub 2022 Jun 29.
- García García E, Gómez Gila AL, Merchante E, Rivero Garvia M, Venegas Moreno E, Soto Moreno A, et al. Endocrine manifestations of central nervous system germ cell tumors in children. Endocrinol Diabetes Nutr (Engl Ed). 2020 Oct;67(8):540-544. doi: 10.1016/j.endinu.2019.11.012. Epub 2020 Mar 18.
- Lo AC, Hodgson D, Dang J, Tyldesley S, Bouffet E, Bartels U, et al. Intracranial Germ Cell Tumors in Adolescents and Young Adults: A 40-Year Multi-Institutional Review of Outcomes. Int J Radiat Oncol Biol Phys. 2020 Feb 1;106(2):269-278. doi: 10.1016/j.ijrobp.2019.10.020. Epub 2019 Oct 22.

