El adolescente con síndrome de Raynaud y vasculitis sistémicas más frecuentes
El adolescente con síndrome de Raynaud y vasculitis sistémicas más frecuentes
M. Garrido Martín(1), C. Udaondo Gascón(2)
(1)Servicio de Pediatría. Complejo Asistencial Universitario de Salamanca. (2)Servicio de Reumatología Pediátrica. Hospital Universitario La Paz. Madrid
Fecha de recepción: 20-12-2023
Fecha de publicación: 31 de marzo 2024
Adolescere 2024; XII (1): 44-57
Resumen
|
El fenómeno de Raynaud (FRy) es una entidad clínica de relativa frecuencia en la adolescencia que, en ocasiones, precede en años al debut de una patología reumatológica subyacente. Su diagnóstico, eminentemente clínico, no exime de la realización de pruebas complementarias y su tratamiento puede ser todo un reto, especialmente si acompaña de ulceración digital o isquemia. La vasculitis se define como una inflamación de la pared de los vasos sanguíneos. Engloban un grupo heterogéneo de enfermedades y en general, su incidencia es baja en edad pediátrica, siendo la vasculitis por IgA o Púrpura de Schönlein-Henoch y la Enfermedad de Kawasaki las más frecuentes. La adolescencia puede ser la edad de inicio de otras vasculitis como las vasculitis ANCA positivas. La sospecha de vasculitis es clínica, debemos pensar en ellas ante síntomas como la fiebre prolongada de causa no infecciosa, la aparición de lesiones cutáneas sugestivas o la elevación de parámetros inflamatorios. La anatomía patológica sigue siendo el gold estándar para su diagnóstico. El tratamiento será específico de cada entidad y el pronóstico variable en función del tipo de vasculitis ante el que nos encontremos. Palabras clave: Raynaud; Acrocianosis; Conectivopatías; Capilaroscopia; Vasculitis; Púrpura de Schönlein-Henoch; Kawasaki; ANCA; Pediatría. |
Abstract
|
Raynaud's phenomenon (RP) is a relatively frequent clinical entity in adolescence, that sometimes precedes in years the onset of an underlying rheumatological pathology. Its diagnosis, eminently clinical, does not exempt the performance of complementary tests and its treatment can be a challenge, especially when it has progressed to digital ulceration or ischemia. Vasculitis is defined as an inflammation of the blood vessel wall, comprising a heterogeneous group of diseases. Their incidence is low in the pediatric age, with IgA vasculitis or Schönlein-Henoch purpura and Kawasaki disease being the most frequent. Adolescence may be the age of onset of other vasculitis such as ANCA-positive vasculitis. The suspicion of vasculitis is clinical, being considered in the face of symptoms such as prolonged fever of non-infectious cause, the appearance of suggestive skin lesions or the elevation of inflammatory parameters. Histology remains the gold standard for diagnosis. The treatment will be specific to each entity and the prognosis will vary depending on the type of vasculitis identified. Key words: Raynaud; Acrocyanosis; Connective tissue diseases; Capillaroscopy; Vasculitis; Henoch-Schönlein purpura; Kawasaki; ANCA; Pediatrics. |
FENÓMENO DE RAYNAUD
Introducción
El fenómeno de Raynaud (FRy) se produce por una respuesta vascular exagerada ante un estímulo externo, que condiciona la disminución de la circulación sanguínea a los dedos de forma reversible. El desencadenante más común es la exposición a temperaturas bajas, aunque también puede producirse tras el ejercicio físico o estrés emocional(1). Esto lleva a la constricción anómala de las arterias digitales y las arteriolas cutáneas, produciendo los cambios característicos en la coloración de la piel(2).
El episodio típico de FRy se caracteriza por frialdad cutánea y cambio en la coloración de los dedos en tres fases: palidez, cianosis e hiperemia. La afectación más frecuente son los dedos de las manos, seguida de los pies
Un episodio típico se caracteriza por frialdad cutánea y cambio de coloración en los dedos en tres fases: una primera fase de palidez, una segunda cianótica, y una tercera de hiperemia (Figura 1). Suelen afectarse los dedos de las manos, seguido de los pies, aunque en ocasiones pueden verse involucradas otras regiones acras del cuerpo como las orejas o la nariz(2).
El FRy se clasifica en dos grandes grupos en función de si está o no asociado a una enfermedad de base, es decir, según su etiopatogenia. Distinguimos por tanto entre primario o secundario. Las características clínicas que nos pueden hacer sospechar que el fenómeno de Raynaud es primario o secundario están resumidas en la Tabla I.
El Raynaud primario se origina por cambios funcionales, sin estar asociado a ninguna enfermedad. Una hiperactividad simpática y un desbalance entre sustancias vasodilatadoras y vasoconstrictoras serían los dos principales elementos determinantes de este proceso. Es el más frecuente, en general tiene un curso benigno y no implica daño orgánico.
Por otro lado, en el Raynaud secundario, existe un daño endotelial producido por la enfermedad de base, que lleva a una desregulación entre factores vasodilatadores y vasoconstrictores. Este daño endotelial lleva a una disminución del flujo sanguíneo distal, existe daño orgánico pudiendo producirse úlceras digitales, pérdida de sustancia o gangrena si no se toman las medidas adecuadas. En este caso, sí existe una patología subyacente como podrían ser algunas enfermedades reumáticas autoinmunes (esclerosis sistémicas, lupus eritematoso sistémico, enfermedad mixta del tejido conectivo, síndrome de Sjögren o dermatomiositis), infecciones, enfermedades metabólicas (crioglobulinemia o ateroesclerosis).
Epidemiología
La prevalencia del FRy no está establecida dada la ausencia de pruebas diagnósticas estandarizadas(1). En general, es más común en el sexo femenino, en adolescentes o en adultos jóvenes y en familiares de personas ya afectas por FRy. Además, es más probable en pacientes que viven en climas fríos. Un estudio en adolescentes de 12 a 15 años en Manchester determinó que, en dicha población, la prevalencia alcanzaba el 18 % en las mujeres y el 12 % en los hombres, aumentando estas cifras a medida que aumentaba la edad(3).
Aunque la mayoría de las personas con FRy no tienen ni desarrollarán un trastorno asociado, es importante saber que sí puede ser el primero de los signos de una enfermedad reumatológica subyacente. En enfermedades como la esclerosis sistémica o la enfermedad mixta del tejido conectivo, el FRy es la primera manifestación clínica en el 80-90 % de los niños afectados(2).
Entre los factores asociados con FRy se encuentran los tratamientos estimulantes del sistema nervioso central para el TDAH
Entre los factores asociados al FRy, se encuentran algunos fármacos entre los que cabe destacar por su frecuencia los tratamientos estimulantes del sistema nervioso central (SNC) para el trastorno de déficit de atención e hiperactividad (TDAH)(4).
Clínica
Como ya se ha mencionado, el FRy se atribuye a una disminución de flujo sanguíneo, esto conlleva una palidez cutánea delimitada al área menos vascularizada. Típicamente empieza en un único dedo y se extiende posteriormente a otras zonas adyacentes, pudiendo afectar a uno o varios dedos. Los dedos índice, medio y anular son los que se ven afectados con mayor frecuencia.
Tras esta primera fase de palidez, la piel se vuelve cianótica debido a la hipoxia tisular. Posteriormente se produce la fase de recalentamiento que conlleva una hiperemia de la región afectada; los dedos pueden hincharse o picar. Tiene un inicio y fin bruscos, y el área de cambio de coloración suele estar bien delimitada. No es infrecuente en pediatría que una de las tres fases esté ausente, tratándose de Raynaud bifásicos.
Como consecuencia de la disminución de flujo sanguíneo, el paciente puede sentir hormigueo, entumecimiento, torpeza y dolor en los dedos. En los casos leves, el recalentamiento revierte la sintomatología. En algunos casos de FRy más severo, la isquemia tisular puede provocar nódulos subcutáneos rojos y dolorosos (perniosis o sabañones) y descamación o ulceración de la piel afectada, así como lesiones vasculíticas.
Otra entidad frecuente es la acrocianosis, una respuesta fisiológica al frío, que cursa con frialdad y cambios de coloración simétricos en manos y pies, afectando también al dorso de las manos
Durante la infancia, es importante diferenciar el FRy de otra entidad relativamente frecuente como es la acrocianosis, una respuesta fisiológica al frío, frecuente tanto en lactantes y niños pequeños como en adolescentes, especialmente con un bajo IMC(1). En este caso, se produce una frialdad y cambios de coloración simétricos en manos y pies simulando un FRy, pero afectando también al dorso de la mano, persistentes, mal delimitados, no dolorosos y sin palidez previa. La perniosis puede aparecer en ausencia de fenómeno de Raynaud, y se incluye dentro de su diagnóstico diferencial. Otras entidades incluidas dentro del diagnóstico diferencial son la eritromelalgia (episodios paroxísticos de vasodilatación con eritema y dolor en regiones acras en respuesta al calor) o la dermatosis o queratodermia acuagénica (aparición de placas de color blanco en las palmas y las plantas en respuesta al agua).
Diagnóstico
El diagnóstico del FRy es predominantemente clínico, no hay pruebas complementarias que lo permitan diagnosticar y, sin embargo, es de gran ayuda que las familias nos muestren fotografías del cambio de coloración para poder confirmarlo(1).
Lo más importante es discernir si nos encontramos ante un Raynaud primario o secundario, ya que como se ha mencionado previamente, el fenómeno de Raynaud puede ser la primera manifestación de una enfermedad reumática como las conectivopatías (esclerosis sistémica, lupus, dermatomiositis, enfermedad mixta del tejido conectivo), cuyo diagnóstico y tratamiento precoz pueden mejorar su pronóstico. Existen otras causas que pueden asociarse al fenómeno de Raynaud (Tabla II).
La herramienta fundamental es una exhaustiva anamnesis y una concienzuda exploración física. Dentro de la anamnesis, deberemos recoger los siguientes datos:
- Antecedentes personales y familiares generales.
- Antecedentes personales y familiares de enfermedades reumatológicas.
- Frecuencia, gravedad y duración de los ataques. Patrón del color.
- Dedos involucrados, grados de afectación y simetría, así como presencia de úlceras digitales. Progresión.
- Posibles desencadenantes y estacionalidad si la hubiese.
- Síntomas asociados.
- Clínica asociada que sugiera una enfermedad del tejido conectivo: fiebre de causa no infecciosa, fatiga, erupción cutánea, fotosensibilidad, rigidez matutina, artralgias, mialgias, disfagia, úlceras orales.
En cuanto a la exploración física, esta deberá ser sistemática y general, para evitar pasar por alto algún signo propio de enfermedad sistémica subyacente.
Las pruebas complementarias incluyen los anticuerpos antinucleares y la capilaroscopia periungueal (visualiza las asas capilares del lecho ungueal)
Como pruebas complementarias, tanto los anticuerpos antinucleares como los hallazgos de la capilaroscopia periungueal pueden ayudarnos a diferenciar a los pacientes con Raynaud primario de aquellos con Raynaud secundario.
La capilaroscopia es una técnica no invasiva que nos permite visualizar las asas capilares del lecho ungueal, para evaluar su densidad, morfología o flujo capilar, visualizar el plexo venoso y el área pericapilar. Se puede realizar con un dermatoscopio o un capilaroscopio, según disponibilidad. La presencia de megacapilares, capilares muy dilatados, telangiectasias y una escasez o pérdida relativa de asas capilares sugieren la presencia de una conectivopatía. Estos hallazgos en un paciente con FRy deben llevar a una investigación más exhaustiva para descartar una posible enfermedad reumática subyacente (Figura 2).
En relación con las pruebas de laboratorio, pueden ser de utilidad para definir si existe o no un trastorno asociado al FRy. Ante el diagnóstico provisional de Raynaud primario deben realizarse al menos dos determinaciones de anticuerpos antinucleares (ANA), con una separación mínima de 3 meses. La presencia de títulos altos nos tiene que hacer pensar en que podríamos encontrarnos ante un Raynaud secundario. Ante la duda, si encontramos unos anticuerpos ANA positivos, se deberían de solicitar como cribado anticuerpos específicos de enfermedades del tejido conectivo como anticuerpos anti-centrómero, anti-DNA y ENA (anti-SCL 70, anti-Ro, anti-La, RNP, Sm).
La combinación en un paciente con FRy de ANA positivos junto con la alteración en la capilaroscopia tiene una fuerte correlación con el FRy secundario.
Los pacientes con FRy con datos de Raynaud secundario (ver Tabla I) deben ser derivados a una unidad de Reumatología para su seguimiento, debido a la posibilidad de una enfermedad subyacente, y su tratamiento específico.
Tratamiento
En pacientes con FRy secundario suelen requerir fármacos para un adecuado control sintomático, y evitar el posible daño orgánico debido a la isquemia
El manejo terapéutico del FRy va a depender de si este es primario o secundario ya que habitualmente los pacientes con FRy primario presentan una clínica más leve, aunque su calidad de vida puede verse afectada por la necesidad de evitar el frío. Sin embargo, los pacientes con un FRy secundario suelen requerir fármacos de forma más frecuente para un adecuado control sintomático y evitar el posible daño orgánico debido a la isquemia.
Vamos a distinguir por tanto entre medidas generales (Tabla III), útiles para todos los pacientes independientemente del tipo de Raynaud que padezcan y su gravedad, y medidas farmacológicas, que se emplearán en aquellos pacientes que así lo requieran.
Medidas no farmacológicas
Los pacientes deben evitar o limitar la exposición al frío y las bebidas estimulantes: cafeína, teína, chocolate y tabaco
Los pacientes deberán evitar o limitar la exposición al frío, deberán siempre ir bien abrigados y llevar guantes cuando así lo requieran las condiciones térmicas. Deberán de evitar bebidas estimulantes que contengan cafeína, teína, así como el chocolate y el tabaco. En la mayoría de los pacientes, sobre todo en aquellos afectos de Raynaud primario, estas medidas serán suficientes para combatir las crisis. En aquellos pacientes que reciban tratamiento farmacológico para el TDAH, además de realizar las medidas no farmacológicas, se recomienda individualizar en cada caso la necesidad de ajustar la dosis o realizar cambios en el tratamiento del TDAH en función de la afectación.
Medidas farmacológicas
Cuando no haya una adecuada respuesta a las medidas generales o ya haya daño secundario a la isquemia se ha de iniciar tratamiento farmacológico, entre estas herramientas terapéuticas orales encontramos:
- Bloqueadores de los canales de calcio: nifedipino y amlodipino.
- Inhibidores de la fosfodiesterasa 5: sildenafilo y tadalafilo.
- Bloqueadores del receptor de angiotensina tipo II: losartan.
- Bloqueadores de los receptores de endotelina 1: bosentan.
- Metilxantinas.
- Bloqueador selectivo de los receptores alfa – 1: prazosina.
- Inhibidor selectivo de la recaptación de serotonina: fluoxetina.
Algunos de estos fármacos se pueden combinar para conseguir el efecto terapéutico que deseamos. Del mismo modo se pueden suspender temporalmente en aquellas épocas en las que la sintomatología mejore.
Por otro lado, también contamos con vasodilatadores tópicos (trinitrato de glicerina, nifedipino tópico) que pueden ser de utilidad al limitar los efectos sistémicos.
En caso de una importante severidad de las lesiones, se puede usar un análogo de la prostaciclina, administrado por vía intravenosa que se considera una terapia de segunda línea, y a menudo en combinación con otras terapias vasodilatadoras. Otro aspecto para tener en cuenta es la utilización concomitante de agentes antiagregantes como el ácido acetilsalicílico o terapia anticoagulante en caso de sospechar la presencia de trombosis asociada.
En último lugar hemos de tener en cuenta de que, si hay una úlcera franca, deberíamos asociar antibioterapia sistémica para evitar la sobreinfección.
Inyección de toxina botulínica
La inyección de toxina botulínica se ha utilizado en Raynaud para disminuir la vasoconstricción periférica. Se utiliza como tratamiento sintomático y se puede repetir cada 6-12 meses en función de cada caso(5).
VASCULITIS EN PEDIATRÍA
Introducción
El término vasculitis reúne a un grupo heterogéneo de enfermedades definidas por la presencia de inflamación en la pared de un vaso sanguíneo, lo cual puede asociar lesiones tisulares directas o secundarias al daño vascular. La inflamación puede ocurrir como un proceso primario o secundario a una enfermedad subyacente. La clínica de las vasculitis va a depender del tipo, localización y tamaño del vaso afecto.
La vasculitis mediada por Inmunoglobulina A o Púrpura de Schönlein-Henoch, seguida de la enfermedad de Kawasaki, son casi exclusivas de la edad pediátrica
Las vasculitis, en general, son poco frecuentes en la edad pediátrica. Se estima una incidencia de vasculitis primarias en pediatría de unos 50 casos/100.000 niños/año, siendo la vasculitis mediada por Inmunoglobulina A o Púrpura de Schönlein-Henoch (PSH), seguida por la enfermedad de Kawasaki (EK), casi exclusivas de la edad pediátrica.
La clasificación de las vasculitis está recogida en la Tabla IV, con criterios establecidos en 2008 y validados por la Liga Europea contra el Reumatismo/Sociedad Europea de Reumatología Pediátrica/ Organización Internacional de Ensayos en reumatología Pediátrica (EULAR/PRINTO/PRES).
Diagnóstico general de las vasculitis
Hemos de sospechar una vasculitis ante la presencia de ciertos hallazgos clínicos como: fiebre prolongada de origen desconocido, lesiones cutáneas sugestivas y afectación renal, pulmonar o cardiovascular de causa desconocida, junto con afectación sistémica, neuropatía periférica o afectación del SNC, serositis o artritis, o elevación de parámetros inflamatorios en la analítica.
El diagnóstico diferencial incluye enfermedades infecciosas, reacción a fármacos, enfermedades oncológicas, otras enfermedades autoinmunes o síndromes autoinflamatorios o incluso vasculopatías no inflamatorias. Se debe tener en cuenta que los niños poseen una mayor susceptibilidad a la inflamación vascular ante un fenómeno externo que los adultos.
En cuanto a exámenes complementarios, algunas pruebas de imagen pueden ser de gran utilidad para visualizar vasculitis de mediano y gran vaso, aunque el gold estándar para su diagnóstico sigue siendo la demostración de inflamación vascular mediante anatomía patológica. En el caso de la Púrpura de Schönlein-Henoch y la enfermedad de Kawasaki, el diagnóstico es fundamentalmente clínico y salvo excepciones no será necesaria la realización de pruebas diagnósticas más agresivas como la biopsia. A lo largo del siguiente artículo se van a explicar algunas de las vasculitis más frecuentes en pediatría.
Vasculitis por IgA o púrpura de Schönlein-Henoch
La vasculitis por IgA (VIgA) clásicamente conocida como Púrpura de Schönlein-Henoch (PSH) es la vasculitis más frecuente en la infancia y, prácticamente, exclusiva de esta franja de edad (el 90 % de los casos ocurren en edad pediátrica). Se estima una incidencia de 326,7 casos por 100.000 habitantes(7).
La edad de presentación de VIgA o PSH es de los 3 a los 15 años de edad, y es más común en varones. Hasta en el 50% de los casos está precedida de una infección de vías respiratorias superiores
Ocurre generalmente entre los 3-15 años de edad y es más común en varones.
Se trata de una vasculitis de pequeño vaso que afecta fundamentalmente a capilares, vénulas y arteriolas. Se caracteriza por el fenómeno de leucocitoclasia y el depósito de inmunocomplejos IgA en la pared vascular(1).
Más frecuente en los meses de enero a marzo, hasta el 50 % de los casos está precedido de una infección de las vías respiratorias superiores. Agentes etiológicos como el estreptococo del grupo A betahemolítico, así como parvovirus, virus de la hepatitis B, virus de la hepatitis C, adenovirus, estafilococo aureus o micoplasma se han visto involucrados en su etiopatogenia.
Las vacunas y las picaduras de insectos son también agentes desencadenantes de esta vasculitis(9).
Para desarrollar una VIgA es preciso una predisposición genética individual sobre la cual incide un factor ambiental. Esto, conduce a la formación de inmunocomplejos que se unen a la pared del vaso generando una respuesta inflamatoria local que desencadena el daño orgánico(1,9).
Clásicamente se ha descrito una tétrada de signos clínicos que caracteriza la PSH:
- Púrpura palpable en ausencia de trombocitopenia y coagulopatía
- Artralgias
- Dolor abdominal
- Afectación renal
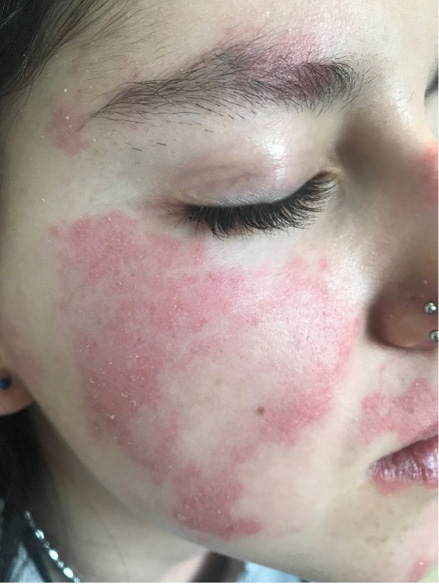
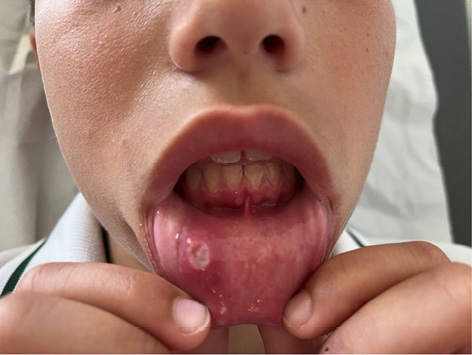
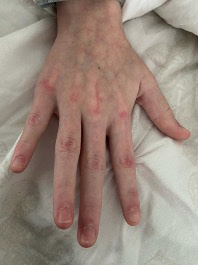
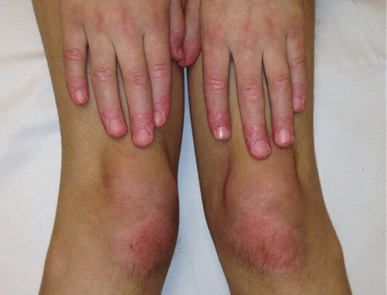
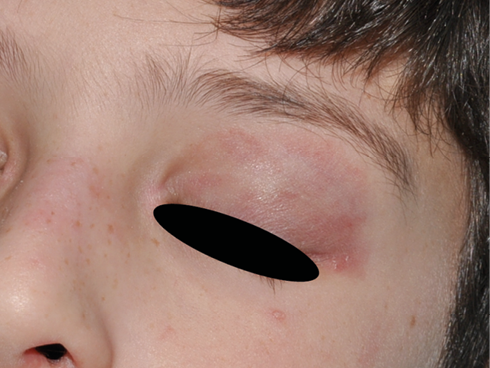
Las lesiones cutáneas aparecen aproximadamente en el 75 % de los pacientes. Las más típicas son las petequias y la púrpura palpable, aunque también puede aparecer un exantema maculopapular o urticarial. Estas lesiones pueden ser pruriginosas pero rara vez son dolorosas. Típicamente aparecen en zonas declives o de presión, de ahí que lo más frecuente sea que aparezcan en extremidades inferiores y glúteos, generalmente de una forma simétrica. A medida que desaparecen dejan pequeñas máculas hiperpigmentadas secundarias a los depósitos de hemosiderina. En niños más pequeños, acompañando a las lesiones puede aparecer edema localizado en cuero cabelludo, manos y pies, cara, escroto o periné.
En relación con las manifestaciones articulares, aparecen entre el 50-80 % de los casos. Las articulaciones más afectas son grandes articulaciones de extremidades inferiores como rodillas y tobillos. Es importante resaltar que suelen asociar inflamación periarticular sin derrame, ni limitación de la movilidad. Las artralgias se caracterizan por ser transitorias y no migratorias.
Las manifestaciones gastrointestinales, suelen aparecer en la primera semanas tras el inicio de las manifestaciones cutáneas. El síntoma más frecuente es el dolor cólico leve-moderado y puede acompañarse de algún vómito. Dentro de las complicaciones digestivas, la invaginación íleo-ileal es la más frecuente. Otro aspecto a señalar es que no es extraño la aparición de sangre oculta en heces.
La afectación renal aparece en los dos primeros meses tras la erupción cutánea, y consiste en una glomerulonefritis en la mayoría de las veces leve y autolimitada
Por último, la afectación renal, que siendo la manifestación más infrecuente de la tétradas es la que marca el pronóstico a largo plazo de esta enfermedad. Puede aparecen en los 2 primeros meses tras la erupción cutánea y consiste en una glomerulonefritis. En la mayoría de los casos son leves y autolimitados. La nefritis en la VIgA generalmente se limita a alteraciones en el contenido de la orina, siendo la microhematuria la más frecuente de ellas y raramente evoluciona a insuficiencia renal. Sin embargo, en aquellos pacientes que desarrollan un síndrome nefrítico o nefrótico, la probabilidad de desarrollar un daño renal crónico a largo plazo aumenta considerablemente. De esto se deduce la importancia de hacer un adecuado seguimiento de la función renal en los pacientes diagnosticados de PSH, al menos durante los primeros 6-12 meses desde el debut(8,9).
No existe una única prueba que permita realizar el diagnóstico, sino que este se basa en hallazgos clínicos.
Como ya se ha mencionado, el gold estándar para el diagnóstico de las vasculitis es la biopsia, aunque rara vez se usa en el caso de la PSH si la clínica es sugestiva. El hallazgo típico de la anatomía patológica es el de una vasculitis leucocitoclástica con depósitos de IgA en la inmunofluorescencia. Sin embargo, la ausencia de estos inmunocomplejos, a pesar de su especificidad, no descarta el diagnóstico de VIgA(8).
Las pruebas de laboratorio las realizaremos con el fin de establecer un diagnóstico diferencial con otras enfermedades y para objetivar el daño orgánico secundario a la propia vasculitis.
En 2019 el grupo europeo Single Hub and Access point for paediatric Rheumatology in Europe (SHARE), publicó 7 recomendaciones para el diagnóstico(8).
Estará indicado realizar una analítica (descartar trombopenia o alteraciones de la coagulación), un test rápido o cultivo faríngeo (descartar estreptococo) y sedimento de orina al debut y seguimiento
A modo de resumen, estará indicado realizar una analítica en todos los pacientes para descartar una trombopenia o una alteración de la coagulación. Se debe realizar un test de diagnóstico rápido o cultivo faríngeo para descartar la infección concomitante de estreptococo, dada su asociación previamente mencionada, y está recomendado realizar al debut y a lo largo del seguimiento sistemático, sedimento de orina para descartar alteraciones a nivel renal.
Solo en casos de presentación atípica estaría indicada la biopsia cutánea con el fin de evidenciar los inmunocomplejos IgA. En caso de afectación renal precoz y grave, estaría indicada la realización de una biopsia renal.
En cuanto al tratamiento, en la mayoría de las ocasiones con medidas higiénico-dietéticas es suficiente. Se ha de garantizar una adecuada hidratación, dieta blanda, reposo y elevación de las extremidades si hay edema. Ante la existencia de dolor, podemos utilizar alguna medidas farmacológicas como son(10):
- Antiinflamatorios no esteroideos (AINEs), contraindicados en caso de hemorragia digestiva o afectación renal.
- Corticoides: se recomienda su uso en caso de afectación articular, testicular, gastrointestinal y si hay evidencias de afectación del sistema nervioso central (documento de consenso SHARE 2019). Con respecto a la clínica digestiva, disminuyen el dolor y el riesgo de invaginación, pero no se ha demostrado que su uso disminuya el riesgo de daño renal.
Por último, mencionar que, en caso de afectación renal grave, se debe considerar el tratamiento con inhibidores de la enzima convertidora de la angiotensina (IECAS), antagonistas del receptor de la angiotensina II (ARA II) o inmunosupresores como la azatioprina, el micofenolato de mofetilo, o si fuera necesario, bolos de metilprednisolona junto con ciclofosfamida.
En general, la VIgA/PSH es una enfermedad autolimitada (sólo un tercio de los pacientes pueden presentar recurrencias) de buen pronóstico, en donde la afectación renal es el principal limitante del curso benigno de esta entidad.
Enfermedad de Kawasaki
La EK es una vasculitis aguda febril que se caracteriza por fiebre, conjuntivitis bilateral, eritema labial y oral, cambios en las extremidades y adenopatías laterocervicales
La enfermedad de Kawasaki (EK) es una vasculitis aguda febril, que afecta a vasos medianos y que conlleva complicaciones potencialmente graves al tener cierta predilección por las arterias coronarias.
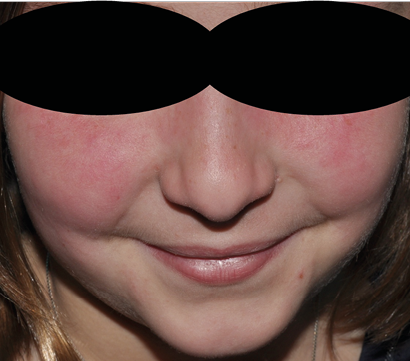
Es la segunda vasculitis más frecuente de la infancia y típicamente se ha descrito en niños asiáticos, menores de 5 años y varones. Se caracteriza por la aparición de fiebre, conjuntivitis bilateral no purulenta, eritema labial y oral, cambios en las extremidades y adenopatías laterocervicales.
Su incidencia es mayor en meses de invierno y primavera y en determinadas regiones del planeta. En Japón se estima una incidencia de 330 por cada 100.000 habitantes, frente a los entre 5,4 y 15 afectos por 100.000 en Europa(7⁾. La afectación de la vascularización coronaria, la ha convertido en la causa más frecuente de enfermedad cardiaca adquirida en niños en países desarrollados.
En cuanto a su etiología, al igual que la PSH u otras vasculitis, se sospecha que sobre una predisposición genética individual actúa un agente externo desencadenando la cascada inflamatoria.
El cuadro clínico se desarrolla en tres fases:
- Periodo febril agudo: los primeros 10 días
- Periodo subagudo: con una duración de entre 2 y 4 semanas
- Periodo de convalecencia: duración variable, es el periodo en el que se resuelve la clínica
Las manifestaciones clínicas más frecuentes, como ya hemos mencionado son: la fiebre, se pueden alcanzar elevadas temperaturas y responde parcialmente a antitérmicos. La conjuntivitis que se caracteriza por ser bilateral y por no acompañarse de secreción purulenta. Las alteraciones bucales, entre las que destaca la afectación labial con marcada xerosis y aparición de grietas. El exantema que suele durar lo mismo que la fiebre, no se acompaña de vesículas ni costras y que afecta característicamente al periné. La afectación en extremidades, suelen evolucionar según el curso de la enfermedad; inicialmente consiste en eritema palmar y plantar, asociado o no a edema, que en la fase de convalecencia da lugar a una descamación periungueal.
Otra manifestación frecuente son las adenopatías, suele ser única, submandibular, indurada y dolorosa de hasta 1,5 cm de diámetro. En último lugar, y la más relevante por ser el mayor determinante pronóstico, la afectación cardiaca que puede ser muy variable, desde pequeñas alteraciones en el electrocardiograma (ECG) sin relevancia clínica hasta pericarditis, endocarditis, miocarditis o aneurismas coronarios.
La afectación cardíaca en la EK es variable: alteraciones del ECG hasta pericarditis, endocarditis, miocarditis o aneurismas coronarios
Dada la relevancia de las manifestaciones cardiacas, no pueden pasar inadvertidas y se han de llevar a cabo una serie de pruebas complementarias que permitan su identificación.
Para el diagnóstico, se han desarrollado una serie de criterios clínicos (Tabla V); en base a los mismos distinguimos dos entidades:
- Enfermedad de Kawasaki completa: cuando se junta el criterio de la fiebre más otros 4 criterios clínicos o fiebre junto a 3 criterios, siempre y cuando el paciente también presente afectación cardiaca compatible.
- Enfermedad de Kawasaki incompleta: cuando se cumple el criterio principal de fiebre con algún otro criterio clínico, habiendo excluido otras enfermedades, pero sin cumplir los criterios necesarios para la EK completa. También podemos hablar de EK incompleta cuando se cumplen todos los criterios menos la duración de la fiebre.
Por último, hablamos de Enfermedad de Kawasaki atípica cuando nos encontramos ante un paciente que presenta manifestaciones clínicas que clásicamente no se observan en la EK.
Como ya se ha mencionado, la relevancia de un adecuado diagnóstico de esta entidad se basa en las lesiones cardiacas que puede llegar a producir, es por eso que se han establecido una serie de criterios cuya presencia se correlacione con la probabilidad de desarrollar aneurismas coronarios(9):
- Edad menor de 12 meses
- Hematocrito > 35 % o descenso paulatino desde el diagnóstico
- Plaquetas < 300.000/mcL
- Hiponatremia < 133 mmol/L
- Elevación de aspartato aminotransferasa (AST) > 100 UI/L
- Elevación de la Proteína C reactiva (PCR) > 200 mg/L
- Albúmina < 35 g/L
- Neutrofilia > 80 %
- Administración de inmunoglobulina intravenosa (IGIV) después del 10º día de fiebre
- Presencia de afectación coronaria al diagnóstico (dilatación o aneurisma, no hiperrefringencia coronaria)
- EK recurrente
- Shock o presencia de síndrome de activación macrofágica.
Cualquier paciente que presente al menos uno de los criterios aquí citados se considerará un paciente de alto riesgo. Clasificar los pacientes en función del riesgo nos va a ser útil a la hora de establecer un tratamiento adecuado, ya que seremos más agresivos con aquellos pacientes que hayamos considerado de alto riesgo.
Dentro de los tratamientos que disponemos caben destacar los siguientes:
- Inmunoglobulinas intravenosas (IGIV), reducen la severidad y frecuencia de los aneurismas coronarios, su uso está indicado en los primeros 10 días desde el inicio del proceso.
- Antiinflamatorios no esteroideos: mientras dure la fiebre, se puede usar tanto ibuprofeno como ácido acetilsalicílico (AAS).
- Corticoides: asociar bolos de metilprednisolona a las IGIV en pacientes de alto riesgo.
- Ácido acetilsalicílico: a dosis antiagregantes desde el fin de la fiebre.
- Inhibidor de la bomba de protones para protección gástrica
Es importante hacer un diagnóstico precoz, una estratificación adecuada del riesgo y un correcto tratamiento, para disminuir en lo posible las secuelas cardíacas
El pronóstico de la EK está claramente limitado por la afectación cardiaca, la cual ocurre hasta en el 20 % de aquellos pacientes que no han sido tratados adecuadamente. De ahí la importancia de un diagnóstico precoz, de una apropiada estratificación del riesgo y de un correcto tratamiento para disminuir en el mayor grado posible las secuelas cardiacas en estos pacientes.
Vasculitis asociadas a anticuerpos antineutrófilos
Este grupo de vasculitis denominadas vasculitis asociadas a anticuerpos antineutrófilos (VAA) se caracterizan por afectar a vasos de pequeño calibre y acompañarse de anticuerpos anticitoplasma de neutrófilo (ANCA). Afectan principalmente a la vía aérea superior e inferior, así como al riñón(12).
Clásicamente se han diferenciado dentro de este grupo de vasculitis tres entidades:
- Granulomatosis con poliangeítis (GPA) o Granulomatosis de Wegener.
- Granulomatosis eosinofílica con poliangeítis (GEPA) o síndrome de Churg-Strauss.
- Poliangeítis microscópica.
Cada una de estas tres entidades tienen sus propias manifestaciones clínicas, forma de presentación y características histológicas.
A diferencia de las entidades previamente descritas en este artículo, las VAA son entidades poco frecuentes en la infancia, y cuando aparecen suelen afectar típicamente a niñas en las segunda década de la vida, entre los 11 y los 14 años.
La etiología de las VAA es desconocida, se cree que la actuación de diferentes agentes sobre pacientes genéticamente predispuestos desencadena la síntesis de ANCA y la formación de granulomas.
La etiología de la VAA es desconocida y las manifestaciones clínicas son muy inespecíficas. Hasta un 50% debutan con un síndrome constitucional que precede al resto de los síntomas
Las manifestaciones clínicas son muy inespecíficas ya que las VAA suelen afectar a diferentes órganos y sistemas, dando lugar a clínica muy variada. Hasta un 50 % de los pacientes debutan con un síndrome constitucional que precede al resto de los síntomas(12).
La GPA se caracteriza fundamentalmente por la afectación del tracto respiratorio superior con afectación de la nariz a la que puede llegar a deformar (deformidad en silla de montar). También es frecuente la afectación pulmonar en forma de hemorragia, pleuritis o afectación intersticial. El daño renal ocurre hasta en el 80 % de los casos y es muy variable; abarca desde anormalidades en la orina hasta daño renal crónico.
En el caso de la GEPA, los síntomas más típicos son los alérgicos acompañados de eosinofilia sistémica y tisular. Además, la afectación cutánea puede apreciarse en casi la totalidad de estos pacientes. Es muy importante también, señalar la afectación cardiaca que se presenta hasta en el 50 % de los casos y es más grave que en los adultos.
Por último, la PAM es la VAA más grave, ya que prácticamente el 100 % de los pacientes va a desarrollar afectación renal. Puede, además, acompañarse de manifestaciones a nivel gastrointestinal, musculoesquelética, cutánea o pulmonar, así como, afectación del sistema nervioso central.
El diagnóstico diferencial hay que hacerlo con infecciones, neoplasias, otras vasculitis y otras enfermedades autoinmunes
Para el diagnóstico de estas entidades es preciso aunar características clínicas, marcadores serológicos y hallazgos anatomopatológicos. El diagnóstico diferencial suele ser con infecciones, neoplasias, otras vasculitis y otras enfermedades autoinmunes.
En cuanto al tratamiento, destacar que suele constar de dos fases, una inicial o de inducción que busca la remisión rápida en estos pacientes y posteriormente, una segunda o de mantenimiento para garantizar esa remisión y evitar la recaída(1,12).
El pronóstico va a depender no solo del daño orgánico ocasionado por la enfermedad sino también por la toxicidad de los fármacos utilizados para conseguir la remisión; siendo la afectación renal el elemento más limitante.
Tablas y figuras
Tabla I. Características clínicas del Raynaud primario y secundario
|
RAYNAUD PRIMARIO |
RAYNAUD SECUNDARIO |
|
Crisis precipitadas por el frío o por emociones. No hay alteraciones vasculares asociadas |
Crisis dolorosas, simétricas y con signos de isquemia distal como úlceras o lesiones vasculíticas |
|
No síntomas sistémicos |
Síntomas sistémicos presentes |
|
Ausencia de necrosis tisular, “pitting” ungueal o gangrena |
Datos de isquemia proximal de los dedos de las manos y de los pies |
|
Estudio por capilaroscopia normal |
Alteración en la capilaroscopia |
|
Anticuerpos antinucleares (ANA) negativos y velocidad de sedimentación (VSG) normal |
Alteración en los valores de laboratorio como positividad de los ANA o elevación de la VSG, disminución del complemento |
Características clínicas que aparecen con más frecuencia en el fenómeno de Raynaud primario o secundario. Adaptado de: Cassidy, James T. Petty, Ross E. Textbook of Pediatric Rheumatology. Ed. Saunders. 6º ed. 2010. ISBN: 978-1-4160-6581-4.
Tabla II. Entidades asociadas con el fenómeno de Raynaud en la edad pediátrica
|
RAYNAUD PRIMARIO |
|
Enfermedades reumatológicas |
|
Esclerosis sistémica Enfermedad mixta del tejido conectivo Lupus eritematoso sistémico Vasculitis Síndrome de Sjögren Síndrome antifosfolípido |
|
Vasoespasmo primario |
|
Migraña |
|
Alteraciones mecánicas u obstructivas |
|
Vasculopatías primarias Traumatismos o congelación recurrente Síndrome de compresión del desfiladero torácico Síndrome del túnel del carpo |
|
Hiperviscosidad sanguínea o desórdenes protrombóticos |
|
Crioglobulinemia Policitemia Enfermedad de células falciformes Trombocitopenia esencial Hiperlipidemia |
|
Alteraciones hormonales |
|
Síndrome carcinoide Feocromocitoma Hipotiroidismo |
|
Infecciones |
|
Parvovirus B19 Helicobacter Pylori |
|
Exposición a drogas o fármacos |
|
Agentes quimioterápicos Agentes vasoactivos (anfetaminas, antihistamínicos, adrenalina) Estimulantes del SNC o fármacos para el TDAH (metilfenidato, dextroanfetamina) Mercurio Cloruro de polivinilo Drogas de recreo (cocaína, LSD, éxtasis) |
|
Otros |
|
Síndrome de Down Malformaciones arteriovenosas Anorexia nerviosa Distrofia simpática refleja |
Entidades asociadas y otras causas del Fenómeno de Raynaud en Pediatría.
Adaptado de: Cassidy, James T. Petty, Ross E. Textbook of Pediatric Rheumatology. Ed Saunders. 6º ed. 2010. ISBN: 978-1-4160-6581-4.
Tabla III. Medidas generales para el manejo del fenómeno de Raynaud
|
Manejo y medidas generales para el manejo terapéutico del fenómeno de Raynaud. Adaptado de: Cassidy, James T. Petty, Ross E. Textbook of Pediatric Rheumatology. Ed Saunders. 6º ed. 2010. ISBN: 978-1-4160-6581-4.
Tabla IV. Clasificación de las vasculitis pediátricas (EULAR/PRES)
|
VASCULITIS PREDOMINANTEMENTE DE VASOS PEQUEÑOS |
|
Granulomatosas |
|
|
No granulomatosas |
|
|
VASCULITIS PREDOMINANTEMENTE DE VASOS MEDIANOS |
|
|
VASCULITIS PREDOMINANTEMENTE DE VASOS GRANDES |
|
|
OTRAS VASCULITIS |
|
Tabla V. Criterios diagnósticos de la enfermedad de Kawasaki
|
CRITERIOS DIAGNÓSTICOS DE ENFERMEDAD DE KAWASAKI COMPLETA |
|
Fiebre de > 5 días de duración y al menos 4 de los siguientes criterios:
|
Adaptado de: Sánchez-Manubens J. Enfermedad de Kawasaki. Protocdiagn ter pediatr. 2020;2:213-224.
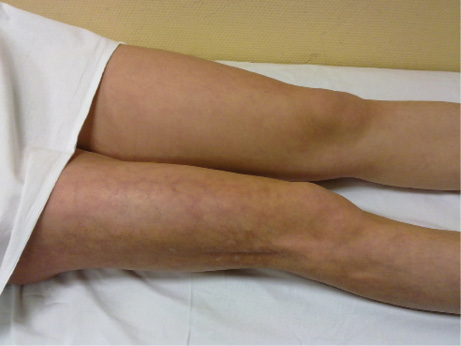
Figura 1. Paciente con fenómeno de Raynaud

En la imagen se puede observar la fase pálida bien delimitada afectando a dedos 2º y 3º.
Cortesía de Dra. C. Udaondo.
Figura 2. Imágenes de capilaroscopias a 50x (DinoLite)
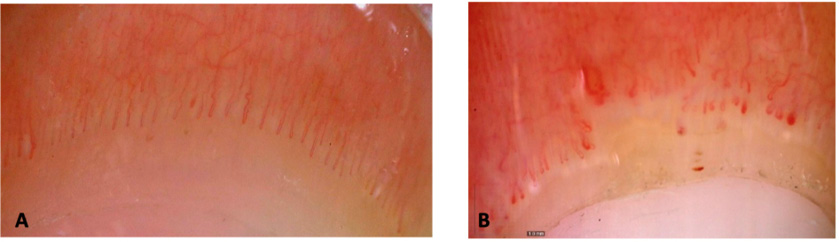
A.Patrón normal. B. Patrón esclerodermiforme: presencia de megacapilares y de áreas avasculares, disminución de la densidad, presencia de microhemorragias. Cortesía de Dra. C. Udaondo.
Bibliografía
- López Robledillo JC, Gámir Gámir ML, eds. Manual SER de diagnóstico y tratamiento en Reumatología Pediátrica. Madrid: Ergons.A.; 2019 p.141-46.
- Cassidy JT. Petty Ross E. Textbook of Pediatric Rheumatology. Ed Saunders. 6º ed. 2010. ISBN: 978-1-4160-6581-4.
- Jones GT, Herrick AL, Woodham SE, Baildam EM, Macfarlane GJ, Silman AJ. Occurrence of Raynaud’s phenomenon in children ages 12-15 years: prevalence and association with other common symptoms. Arthritis Rheum. 2003 Dec;48(12):3518-21. doi: 10.1002/art.11340. PMID: 14674003.
- Iglesias Otero M, Portela Romero M, Bugarín González R, Ventura Victoria M. Methylphenidate and secondary Raynaud’s phenomenon. Semergen (2013) 39(6) 330-334.
- Lawson O, Sisti A, Konofaos P. The Use of Botulinum Toxin in Raynaud Phenomenon: A Comprehensive Literature Review. Ann Plast Surg. 2023 Jul 1;91(1):159-186. doi: 10.1097/SAP.0000000000003603. PMID: 37450876.
- Herrick AL. Evidence-based management of Raynaud’s phenomenon. Ther Adv Musculoskelet Dis. 2017 Dec;9(12):317-329. doi: 10.1177/1759720X17740074. Epub 2017 Nov 20. PMID: 29201156; PMCID: PMC5700788.
- Antón López J, Carriquí Arenas S. Púrpura de Schönlein-Henoch, enfermedad de Kawasaki y otras vasculitis. Pediatr Integral 2022; XXVI (3): 151 – 162.s
- Ozen S, Pistorio A, Iusan SM, Bakkaloglu A, Herlin T, Brik R, et al. EULAR/ PRINTO/PRES criteria for Henoch-Schönlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II: Final classification criteria. Ann RheumDis. 2010; 69: 798-806.
- Borlán Fernández S. Vasculitis por IgA (púrpura de Schönlein-Henoch). Protocdiagn ter pediatr. 2020;2:225-238.
- Ozen S, Marks SD, Brogan P, Groot N, de Graeff N, Avcin T, et al. European consensus-based recommendations for diagnosis and treatment of immunoglobulin A vasculitis-the SHARE initiative. Rheumatology (Oxford). 2019 Sep 1;58(9):1607-1616. doi: 10.1093/rheumatology/kez041. PMID: 30879080.
- Sánchez-Manubens J. Enfermedad de Kawasaki. Protocdiagn ter pediatr. 2020;2:213-224.
- Alcobendas Rueda RM, Remesal Camba A, Fernández Fraga P. Vasculitis asociadas a ANCA positivo. Protoc diagn ter pediatr 2020; 2: 239-248.
- Martini A. Hachulla E. EULAR Textbook on Paediatric Rheumatology. Editorial BMJ. ª Edición Noviembre 2018. ISBN 9780727918833.
No existen conflictos de interés en la realización de este artículo.