Pubertad precoz y retraso puberal
Pubertad precoz y retraso puberal
J. Pozo Román(1,2,3), M. Márquez Rivera(1), M.T. Muñoz Calvo (1,2,3).
(1)Servicios de Pediatría y Endocrinología. Hospital Infantil Universitario Niño Jesús. Instituto de Investigación La Princesa. Madrid. (2)Universidad Autónoma de Madrid. Departamento de Pediatría.(3)CIBER Fisiopatología de la obesidad y nutrición. Instituto de Salud Carlos III. Madrid.
Fecha de recepción: 1 de febrero 2017
Fecha de publicación: 28 de febrero 2017
Adolescere 2017; V (1): 23-49
Resumen
|
La edad en que la pubertad se inicia es muy variable y, en condiciones normales, está influenciada, además de por el sexo, por factores genéticos y ambientales. Su presentación precoz o tardía puede ser una simple variación extrema de la normalidad o el reflejo de alguna de las múltiples patologías que pueden influir o condicionar el momento de su aparición. En cualquier caso, aun en ausencia de patología, el desarrollo de los caracteres sexuales secundarios a una edad “inadecuada” es motivo de preocupación para los padres y los pacientes y puede tener repercusiones negativas en la vida adulta. El pediatra de Atención Primaria es el profesional que se enfrenta inicialmente a este tipo de situaciones y debe ser capaz de realizar un enfoque diagnóstico adecuado, diferenciar aquellas variantes normales del desarrollo de las que no lo son, orientar a los padres y al paciente y, en aquellos casos que considere necesario, derivar al paciente a un servicio de Endocrinología Pediátrica. Palabras clave: Pubertad normal, Pubertad precoz, Pubertad adelantada, Retraso puberal. |
Abstract
|
The age in which puberty initiates varies greatly and, under normal conditions, it is also influenced by gender, genetic and environmental factors. Its early or late presentation may be a simple extreme variation of normality or the manifestation of any of the multiple pathologies that may influence the moment of its appearance. In any case, even in the absence of disease, the development of secondary sexual characteristics at an “inadequate” age is a reason for concern for the parents and the patients and may have negative repercussions in the adult age. The primary healthcare pediatrician is the professional who initially faces this situation and one who should be capable of performing an adequate diagnostic approach, differentiating normal variants of development from those which are not, guiding the parents and patient, and in those cases were considered necessary,referring the patient to a Pediatric Endocrinology service. Key words: Normal puberty; Precocious puberty; Early puberty; Pubertal delay. |
Introducción
Las alteraciones en la cronología de la pubertad pueden determinar trastornos no solo del desarrollo gonadal y genital, sino también, del crecimiento, de la composición y proporciones corporales, así como de los aspectos psicológicos y emocionales propios de la adolescencia.
PUBERTAD PRECOZ (PP)
Concepto
Pubertad precoz es la aparición de los caracteres sexuales secundarios antes de los 8 años en las niñas y de los 9 años en los niños (≈ 2,5-3 SDS por debajo de la edad media para el sexo y población estudiada)
Se define la PP como: la aparición de los caracteres sexuales secundarios antes de los 8 años en las niñas y de los 9 años en los niños (≈ 2,5-3 SDS por debajo de la edad media para el sexo y población estudiada; Figura 1)(1)
Unido al concepto de PP, estaría el de “pubertad adelantada” (PA), como: aquella que se inicia próxima a los límites inferiores de la normalidad. Tampoco existe un consenso internacional que establezca con claridad sus límites, pero podríamos situarlos entre los 8-9 años en las niñas y entre los 9-10 años en los niños. Al igual que la PP, la PA es mucho más frecuente en niñas y se considera habitualmente la expresión de uno de los extremos del rango normal de edad de desarrollo puberal; es decir, una variante de la normalidad, que puede ser familiar (“aceleración constitucional del crecimiento y de la pubertad”; ACCP) o esporádica, pero que no precisaría, en general, tratamiento. Los niños con esta variante se caracterizan, clínicamente, por un patrón de crecimiento y desarrollo característico, que conlleva un crecimiento acelerado a partir de los 1-2 años de vida, con talla prepuberal por encima del carril correspondiente a su talla diana, aceleración de la edad ósea (1-3 SDS por encima de su edad cronológica), inicio puberal a una edad en los límites inferiores del rango normal y finalización temprana del desarrollo puberal y del crecimiento, alcanzando, habitualmente, una talla acorde con su contexto familiar.
El desarrollo puberal precoz acelera el ritmo de crecimiento, pero más aún la maduración ósea; de forma que, aunque estos niños parezcan inicialmente altos, el cierre de los cartílagos de crecimiento y la finalización del crecimiento a una edad temprana conlleva un riesgo de modificación de las proporciones corporales (acortamiento de extremidades) y, especialmente, de pérdida de talla final(2). Esta pérdida puede ser muy variable, pero suele ser tanto mayor cuanto más precoz es el inicio puberal, mayor la edad ósea (EO), mayor el tiempo de evolución y mayor la rapidez de progresión de la EO y del desarrollo puberal. Además, existirían determinadas poblaciones de riesgo para una mayor afectación de la talla, como serían aquellos pacientes con:
— Talla baja en el momento del diagnóstico
— Cuadro sindrómico asociado a talla baja
— Antecedentes de RNPEG (recién nacido pequeño para la edad gestacional)
— Deficiencia asociada de hormona de crecimiento (GH)
— Patología oncológica con radioterapia craneal y, especialmente, cráneo-espinal
— Antecedentes de adopción internacional
Las consecuencias psicosociales y conductuales de presentar una PP han sido poco estudiadas y, además, pocas veces son tenidas en consideración a la hora de valorar a estos pacientes. En las niñas, donde mayoritariamente se han llevado a cabo estos estudios, se han descrito situaciones de estrés y de dificultad de adaptación ante los rápidos cambios físicos y psicológicos que se producen, rechazo por parte de sus compañeros y tendencia al aislamiento y a la depresión. Ambos sexos, pero sobre todo los varones, pueden mostrar un aumento de la líbido, con desarrollo de conductas masturbatorias o sexuales inadecuadas, especialmente si existe un cierto grado de retraso mental que limite su capacidad de control. En las niñas, se ha descrito también un inicio más precoz de las relaciones sexuales e, incluso, un mayor riesgo de abuso sexual. Las pacientes que han tenido una PP muestran en la adolescencia más problemas de conducta, incluso delictiva, menor competencia social y suelen alcanzar niveles educativos más bajos(3). Esta desadaptación social parece limitarse a la adolescencia y desaparece en la edad adulta. En cualquier caso, al igual que ocurre con los riesgos auxológicos, los riesgos psicosociales en un paciente concreto son difíciles de predecir.
Clasificación
La pubertad precoz se clasifica en central, periférica y mixta
El incremento de los esteroides sexuales (ES), que es lo que determina la aparición y desarrollo de los caracteres sexuales secundarios, puede tener distinto origen (tabla I), lo que permite clasificar la PP en:
• PP central (PPC). Conocida, también, como: PP verdadera o PP dependiente de gonadotropinas; ya que, el incremento de ES es el resultado de una reactivación normal, pero precoz, del eje hipotálamo-hipófiso-gonadal (HHG) y, por ello, siempre es isosexual.
• PP periférica (PPP). Conocida, también, como: pseudopubertad precoz o PP independiente de gonadotropinas. La fuente de ES puede ser exógena o endógena, gonadal o extragonadal, pero, en cualquier caso, el desarrollo de los caracteres sexuales secundarios no es la consecuencia de la activación del eje HHG. Dependiendo del ES aumentado (andrógeno o estrógeno) y del sexo del niño, las manifestaciones clínicas puede ser isosexuales (apropiadas al sexo del niño) o contrasexuales (contrarias al sexo del niño).
• PP mixta (PPM). Se la conoce, también, como: PP combinada o PPC secundaria; ya que, resulta de una mezcla o combinación de una PPP y una PPC. La exposición prolongada a ES, provocada por una PPP, aceleraría el crecimiento, la EO y la maduración de los centros hipotalámicos implicados en el inicio del desarrollo puberal, causando la reactivación precoz del eje HHG y el desarrollo secundario de una PPC.
Etiopatogenia y manifestaciones clínicas
Pubertad precoz central (PPC)
La PPC se debe a una reactivación precoz del eje HHG y se caracteriza clínicamente por un desarrollo precoz de los caracteres sexuales secundarios, que siempre son isosexuales y que siguen la secuencia normal de aparición, con aumento de tamaño y de la actividad de las gónadas.
La PPC se caracteriza clínicamente, en las niñas, por la aparición (telarquia) y desarrollo progresivo de la mama antes de los 8 años, que puede ser, inicialmente y durante unos meses, unilateral, y por el incremento del volumen testicular (≥ 4 mL) en los varones antes de los 9 años, que es seguido, habitualmente y en ambos casos, por el desarrollo de la pubarquia y axilarquia. La EO se adelanta, habitualmente más de 2 SDS por encima de la edad cronológica y, en las niñas, la VC experimenta una rápida aceleración (estirón puberal) que coincide o incluso puede preceder a la aparición del botón mamario. En los varones, el estirón puberal es un fenómeno más tardío en el desarrollo puberal (estadio III-IV de Tanner) y no suele coincidir con el incremento inicial del volumen testicular; no obstante, en los casos de PPC, también tiende a adelantarse (finales del estadio II o principio del III)(4).
Es una patología con un claro predominio en niñas, en las que la etiología es habitualmente idiopática; mientras que, en los niños son más frecuentes que en las niñas las causas orgánicas (40-94 % frente a un 10-20 %). Otro factor relacionado con la organicidad es la edad; de forma que, a menor edad de inicio puberal, mayor riesgo de organicidad.
Dentro de las formas de PPC idiopática, alrededor de un 30 % son familiares
Dentro de las formas de PPC idiopática, alrededor de un 30 % son familiares(5). Es probable que la mayoría correspondan a formas más o menos extremas de ACCP; no obstante, se han descrito situaciones de PPC familiares de base monogénica. Las primeras que se describieron fueron mutaciones activadoras en el sistema de las kisspeptinas, un grupo de péptidos que resultan del proceso proteolítico del producto del gen KiSS1 (1q32), secretados por neuronas de los núcleos arcuato y anteroventral del hipotálamo, y que se consideran como unos de los principales mediadores del inicio de la pubertad. Estos péptidos, en condiciones normales, incrementan su secreción antes del inicio de la pubertad y se unen a un receptor ligado a las proteínas G de la membrana de las neuronas productoras de GnRH, denominado GPR54 (GPR54 o KiSS1R, en 19p13.3), incrementando considerablemente la amplitud de los picos secretorios de GnRH. Se han descrito dos mutaciones activadoras en KiSS1, una en homocigosis y otra en heterocigosis, y una mutación activadora en el gen que codifica para su receptor (KiSS1R), todas ellas asociadas a una PPC hereditaria, aunque con expresividad familiar variable(5). Más recientemente, se han descrito en varias familias con PPC familiar, mutaciones en heterocigosis, con pérdida de función en el alelo paterno, en el gen MKRN3 (15q11.2).
Dentro de las causas orgánicas de PPC, la más frecuente es el hamartoma hipotalámico(6), que puede encontrarse en un 2-28 % de los casos de PPC
Dentro de las causas orgánicas de PPC, la más frecuente es el hamartoma hipotalámico(6), que puede encontrarse en un 2-28 % de los casos de PPC. Estos tumores son malformaciones congénitas benignas formadas por tejido nervioso desorganizado en el que se incluyen neuronas productoras de GnRH. Aunque se ha especulado que estas neuronas podrían actuar como un generador ectópico de GnRH, el mecanismo responsable de la PPC no está plenamente aclarado. Típicamente, en la RM craneal, los hamartomas aparecen como una masa pedunculada que cuelga del hipotálamo, entre el tuber cinereum y los cuerpos mamilares, justo detrás del quiasma óptico. Pueden ser asintomáticos o asociarse a: PPC, crisis gelásticas (crisis comiciales de risa inmotivada), epilepsia secundariamente generalizada y alteraciones cognitivas y conductuales. La mayoría de estos tumores no crecen o lo hacen muy lentamente y responden al tratamiento habitual con análogos de GnRH; por lo que, la cirugía no suele estar indicada, salvo que condicionen una epilepsia intratable.
Otras múltiples alteraciones del sistema nervioso central (SNC) como: malformaciones, tumores, gliomas hipotalámicos o del quiasma en la neurofibromatosis tipo I, displasia septo-óptica, mielomeningocele e hidrocefalia, entre otras, pueden provocar una pubertad precoz (tabla I). Se cree que estas lesiones alterarían las señales de inhibición tónica que recibe el hipotálamo, favoreciendo la reactivación del eje HHG.
La irradiación craneal, utilizada en el tratamiento de tumores del SNC o como tratamiento coadyuvante en otras patologías oncológicas, como la leucemia linfoblástica aguda, sobre todo cuando se administra a edades muy precoces, puede predisponer al desarrollo de una PA o PPC. Dosis bajas de radioterapia (18-24 Gy) a menudo se asocian a PPC en niñas; mientras que, dosis por encima de 25 Gy incrementan el riesgo de PPC en ambos sexos, con frecuente asociación a deficiencia de GH, combinación que puede ser especialmente negativa para la talla adulta del paciente. Por encima de los 30 Gy, es frecuente que tras una pubertad temprana/precoz desarrollen una deficiencia de gonadotropinas (hipogonadismo hipogonadotropo). Cuando se administran 50 o más Gy, no suele haber PP, sino una falta de desarrollo puberal secundaria a hipogonadismo hipogonadotropo(6).
Pubertad precoz periférica (PPP)
La pubertad precoz periférica no es una verdadera pubertad, sino una pseudopubertad precoz, y los caracteres sexuales secundarios pueden, no solo perder su secuencia de aparición habitual, sino, en algunos casos, ser contrarios al sexo del paciente (contrasexuales)
Es aquella PP en la que el incremento de ES responsable del desarrollo de los caracteres sexuales secundarios no es debido a una activación prematura del eje HHG. Por tanto, no es una verdadera pubertad, sino una pseudopubertad precoz, y los caracteres sexuales secundarios pueden, no solo perder su secuencia de aparición habitual, sino, en algunos casos, ser contrarios al sexo del paciente (contrasexuales).
Los ES implicados en el desarrollo de una PPP pueden ser andrógenos, estrógenos o una mezcla de ambos(7), y su origen exógeno (fuentes externas alimenticias, tópicas, orales o parenterales) o endógeno (suprarrenal o gonadal). El eje HHG no solo no está activado, sino que suele estar completamente inhibido por el exceso de ES circulantes, a través de los mecanismos normales de retrocontrol negativo. Las manifestaciones clínicas de la PPP dependen, fundamentalmente, del tipo de ES responsable:
• En las niñas, la pérdida de la secuencia normal en la aparición de los caracteres sexuales secundarios supone que, por ejemplo, el primer signo de desarrollo puberal pueda ser una menstruación, sin un claro desarrollo mamario previo ni aceleración del crecimiento. El hallazgo de hiperpigmentación areolar precoz con desarrollo mamario todavía incompleto es sugerente de rápida y marcada elevación de los niveles séricos de estrógenos. La presencia de un sangrado vaginal en ausencia completa de telarquia es más sugerente de una causa local (agresión sexual, cuerpo extraño o tumor vaginal) que de una verdadera PP. Un acné severo, de rápida progresión y, especialmente, el agrandamiento del clítoris (virilización) debe hacernos sospechar la presencia de un tumor productor de andrógenos.
• En el caso de los varones, es muy característico de la PPP, el desarrollo progresivo de signos de virilización, como sería el incremento del tamaño del pene, sin un aumento significativo del tamaño testicular. En algunos casos (testotoxicosis, restos adrenales testiculares, tumores productores de gonadotropina coriónica – HCG -, etc.), el volumen testicular puede incrementarse ligeramente (4-8 mL), pero, en cualquier caso, es un volumen inadecuado para el grado de desarrollo de los caracteres sexuales secundarios. El desarrollo de signos feminizantes (ginecomastia marcada) es excepcional, pero puede producirse en el contexto de exposición a una fuente externa de estrógenos o en raros casos de tumores testiculares (tumor de células de Sertoli asociado al síndrome de Peutz-Jegher) o adrenales productores de estrógenos.
Las causas que pueden determinar una PPP quedan reflejadas en la tabla I. Las dos entidades nosológicas más características responsables de PPP son: el síndrome de McCune-Albright y la testotoxicosis.
El síndrome de McCune-Albright (SMA) es una enfermedad rara, con una prevalencia estimada de 1:100.000-1.000.000, más frecuente en niñas, aunque puede darse en ambos sexos(8). Originalmente, fue definido clínicamente por la tríada de: displasia fibrosa poliostótica, manchas de color “café con leche” de bordes irregulares (“en costa de Maine”) y PPP, debida al desarrollo de quistes ováricos autónomos secretores de estrógenos. Posteriormente, se ha visto que otras endocrinopatías hiperfuncionantes pueden estar presentes, como: hipertiroidismo, gigantismo hipofisario, hipercortisolismo o raquitismo hipofosfatémico, entre otras. Se debe a una mutación activadora postcigótica en el gen de la subunidad alfa de la proteínas G de la membrana (GNAS1; 20q13.32), que se produce temprano en la embriogénesis y determina un mosaicismo que puede afectar, de forma variable, a tejidos endocrinos (gónadas, tiroides, adrenales, hipófisis y paratiroides) y no endocrinos (timo, bazo, páncreas, riñón, corazón, etc.). Debido a ello, la expresividad clínica puede ser, también, muy variable y el diagnóstico, en algunos casos, difícil.
La testotoxicosis o PP familiar del varón es una forma de PPP limitada a los varones, debida a una mutación activadora, de herencia autosómica dominante (esporádica o familiar) en el receptor de LH (LHCGR; 2p16.3), que ocasiona una activación autónoma de las células de Ley-dig. Suele ponerse de manifiesto a los 2-4 años de edad con la aparición de: signos puberales, virilización y aceleración del crecimiento, que conduce a una talla final baja por cierre precoz de los cartílagos de crecimiento. En niñas, estas mutaciones no producen PPP; ya que, es necesaria la presencia de LH y FSH para la producción de estrógenos.
Los quistes foliculares ováricos secretan estrógenos de forma transitoria, lo que puede ocasionar desarrollo mamario y, en ocasiones, cuando la producción estrogénica cae, sangrado vaginal esporádico. Los quistes que producen esta sintomatología suelen ser relativamente grandes y pueden ser recurrentes, lo que puede acelerar la maduración ósea y favorecer el desarrollo de una PA o una PPC secundaria. Además, en los que alcanzan un mayor tamaño, puede favorecerse la torsión ovárica y la necesidad de tratamiento quirúrgico.
La exposición a ES exógenos (disruptores endocrinos) puede determinar una PPP iso o contrasexual, dependiendo de su acción hormonal y del sexo del paciente. Una de las hipótesis propuestas para explicar la alta incidencia de PA/PPC en niñas adoptadas de países en vías de desarrollo sería la de los disruptores endocrinos. En estas niñas se han encontrado frecuentemente niveles elevados de pesticidas derivados del DDT (diclorodifenildicloroetano), productos que siguen utilizándose en estos países como insecticidas en el ámbito rural. Según esta hipótesis, la exposición crónica a la actividad estrogénica de estos productos podría madurar el hipotálamo al tiempo que suprimiría su actividad. La eliminación de la exposición al migrar a países desarrollados, donde estos pesticidas están prohibidos, supondría el incremento de la liberación de GnRH y el inicio de la pubertad.
La exposición a ES exógenos (disruptores endocrinos) puede determinar una PPP iso o contrasexual, dependiendo de su acción hormonal y del sexo del paciente
Tumores ováricos, testiculares o adrenales productores de ES, son causas raras de PPP iso o contrasexual. Los tumores ováricos (sobre todo los de células de la granulosa/teca) frecuentemente causan síntomas locales (dolor, distensión, ascitis, efecto masa, etc.) y suelen secretar estrógenos, aunque en ocasiones producen andrógenos y virilización. El diagnóstico en las formas típicas se basa en el hallazgo ecográfico de una masa sólida o sólido-quística en el ovario,con niveles séricos elevados de estradiol y suprimidos de LH/FSH. Los tumores testiculares más frecuentemente asociados a PPP son los derivados de las células de Leydig, que suelen ser benignos en la infancia y presentarse con signos de virilización, incremento del tamaño de uno de los testículos (nódulo, frecuentemente palpable) y niveles séricos elevados de testosterona. Los tumores adrenales (adenomas y adenocarcinomas) suelen producir andrógenos (virilización) y cortisol (síndrome de Cushing); si bien, excepcionalmente pueden producir también estrógenos y feminización.
Pubertad precoz mixta o combinada
Se habla de PP mixta o combinada, cuando una PPC desencadena secundariamente una PPC.
Niños expuestos a altos niveles séricos de ES como consecuencia de una PPP (hiperplasia suprarrenal congénita pobremente controlada, SMA…) pueden desarrollar una posterior PPC. Se ha especulado que los ES podrían “impregnar” el hipotálamo, causando la maduración del eje HHG y la puesta en marcha de la PPC; o bien que, el hipotálamo, acostumbrado a una inhibición por niveles muy elevados de ES, al disminuir estos como resultado de un tratamiento efectivo de la PPP, se reactivara, poniendo en marcha la PPC. En la mayoría de los casos, la PPC solo se inicia si la EO es superior a los 10 años.
Evaluación diagnóstica
La aparición antes de los 8 años en las niñas y de los 9 años en los niños de los caracteres sexuales secundarios puede ser la manifestación de una PP (central o periférica y de causa orgánica o idiopática), pero con más frecuencia se tratará de una variante normal, benigna y no progresiva, de pubertad, que no requerirá habitualmente tratamiento (algoritmo 1). Por ello, un diagnóstico correcto puede evitar costosos y prolongados tratamientos no exentos de potenciales efectos secundarios. La evaluación diagnóstica de estos pacientes conlleva una anamnesis y exploración completas, con énfasis en determinados aspectos, y un número limitado de pruebas complementarias básicas(1).
Anamnesis detallada
Debe interrogarse sobre la edad y el orden de aparición de los caracteres sexuales secundarios, así como sobre las características de su progresión (rápida, lenta, cíclica). En las niñas, la evolución cíclica de la telarquia es muy sugerente de quistes foliculares recurrentes. La aparición precoz de vello pubiano y/o axilar sin otros signos de virilización (clitoromegalia o aumento del tamaño del pene, hirsutismo, marcada aceleración de la EO, aumento de la masa muscular…) sugiere adrenarquia prematura (variante de la normalidad) y no PP, y puede acompañarse de otros signos de androgenización leve-moderada, como: aceleración de la EO de 1-2 años, olor corporal de características puberales, presencia de comedones o acné leve y aumento de grasa en el pelo. Deben recogerse y llevarse a la gráfica de crecimiento los datos de la evolución de la talla, el peso y, sobre todo, de la VC. Debe interrogarse, también, sobre posibles signos de hipertensión intracraneal (cefalea, vómitos o trastornos visuales) sugerentes de patología orgánica intracraneal. Los antecedentes familiares (padres, hermanos y familiares próximos) de PA o PP, así como de patologías hereditarias que puedan modificar el tempo normal de la pubertad deben ser recogidas (hiperplasia suprarrenal congénita, testotoxicosis, resistencia a glucocorticoides…)
Exploración física
Se deben recoger cuidadosamente los parámetros antropométricos (talla, peso y proporciones corporales), el estadio puberal de Tanner, así como otros datos sugerentes de desarrollo puberal, virilización o feminización: olor corporal, acné, ginecomastia, galactorrea, pigmentación areolar, estrogenización de la mucosa vaginal, leucorrea, volumen y simetría testicular, tamaño del clítoris, etc. Debe recogerse, también, la presencia, localización y características de manchas cutáneas sugerentes de neurofibromatosis o SMA.
Edad ósea (EO)
La edad ósea suele ser la primera prueba complementaria a realizar ante la sospecha de una PP; ya que, en condiciones normales, el grado de desarrollo puberal se correlaciona mejor con la EO que con la edad cronológica
Suele ser la primera prueba complementaria a realizar ante la sospecha de una PP; ya que, en condiciones normales, el grado de desarrollo puberal se correlaciona mejor con la EO (r = 0,82) que con la edad cronológica (r = 0,72). En las PPC idiopáticas, la EO está típicamente acelerada al menos un 20% por encima de la edad cronológica (≈ 2 SDS) y, en el caso de las niñas, próxima, habitualmente, a los 10-11 años. Por el contrario, en las PP de causa orgánica, la aceleración de la EO es más variable y depende de la duración y grado de exposición previa a los ES. También, la EO es útil para realizar predicciones de talla adulta y valorar la posible repercusión de la PP sobre la talla final, tanto en el momento del diagnóstico como a lo largo del seguimiento del paciente; no obstante, hay que tener en consideración que la fiabilidad de estas predicciones, en condiciones normales es escasa y, en los pacientes con PP, cuando la EO está muy acelerada, todavía menor y con tendencia a sobrevalorar las expectativas de talla adulta.
Determinaciones hormonales
La presencia de niveles séricos elevados de estradiol o testosterona sería, lógicamente, lo primero a demostrar ante una sospecha de PP; sin embargo, su determinación puede no ser de mucha utilidad en las fases iniciales de la pubertad, ya que, se sitúan con frecuencia por debajo del límite de detección de los inmunoanálisis convencionales, especialmente en el caso del estradiol.
La determinación en suero de andrógenos suprarrenales o de sus precursores, especialmente: sulfato de dehidroepiandrosterona (SDHEA), ∆4-androstendiona y 17-OH-progesterona, puede ser de utilidad en aquellas situaciones clínicas sugerentes de incremento de andrógenos de posible origen adrenal, como sería el caso de tumores suprarrenales o de la hiperplasia suprarrenal congénita; en este último caso, puede ser necesaria la estimulación previa con ACTH (test de ACTH) para descartar formas de presentación tardía.
La prueba hormonal más importante en el diagnóstico de PP es la determinación de los niveles séricos de gonadotropinas (LH y FSH) tras estímulo con 100 μg/m2 de LHRH (test de LHRH) o con un análogo de GnRH (test de GnRHa)
La prueba hormonal más importante en el diagnóstico de PP es la determinación de los niveles séricos de gonadotropinas (LH y FSH) tras estímulo con 100 µg/m2 de LHRH (test de LHRH) o con un análogo de GnRH (test de GnRHa)(9). Esta prueba nos permitiría distinguir, al menos teóricamente, una PPC (patrón de respuesta puberal: claro incremento de LH y FSH, con predominio de LH: cociente LH/FSH > 1) de una variante de la pubertad no progresiva (patrón de respuesta prepuberal: escaso o nulo incremento de LH y FSH con predominio de FSH: LH/FSH <1) y de una PPP (secreción de LH/FSH inhibida). Con los inmunoanálisis más modernos, el punto de corte para una respuesta prepuberal de LH sería de alrededor de 5 mUI/mL. Por otra parte, en la situación más frecuente en la práctica clínica, como es el caso de las niñas con inicio de desarrollo mamario entre los 7 y 8 años (70 % de las telarquias prematuras), la diferenciación entre una PPC idiopática (susceptible de tratamiento) y una telarquia prematura aislada (variante de la normalidad que no precisa tratamiento) puede ser muy difícil y, a veces, sólo un seguimiento estrecho puede diferenciarlas (tabla II). En estos casos, es frecuente el hallazgo de un patrón de respuesta de LH y FSH a LHRH intermedio entre lo que hemos denominado puberal y prepuberal (incremento moderado en los picos de secreción de LH y FSH con predominio de FSH o solo un claro incremento de FSH); de hecho, algunos autores consideran que estas formas de telarquia prematura de evolución variable (pueden regresar, mantenerse sin progresar o progresar lentamente) serían parte de un continuum en la activación del eje HHG y que entre un 15-20 % de ellas terminan evolucionando hacia una PP.
Pruebas de imagen
Una RM craneal, para evaluar la anatomía de la región hipotálamo-hipofisaria y descartar patología orgánica, debería realizarse si se demuestra una activación precoz del eje HHG, especialmente en varones, donde el riesgo de patología orgánica es claramente superior (40-90 % en varones frente a un 8-33% en niñas)(1). En las niñas, cuando la pubertad se inicia entre los 6 y 8 años y sin sintomatología neurológica, el riesgo de patología orgánica es muy escaso (2-7 %) y se discute la conveniencia de realizar una RM craneal; no obstante, la mayoría de los centros continúa realizándola.
La ecografía abdómino-pélvica puede, además de descartar la presencia de tumoraciones (suprarrenales, ovario, hígado, etc.) responsables de una PPP, permitirnos, en el caso de las niñas, valorar el tamaño ovárico y uterino. Un volumen ovárico (longitud x anchura x altura x 0,5233) de < 1 mL es claramente prepuberal, pero los límites para considerarlo puberal, varían entre 1 y 3 mL, según los autores. La presencia de pequeños quistes (< 9 mm), en ocasiones múltiples (2 a 4), es un hallazgo frecuente y normal en niñas prepuberales (50-80 %). Típicamente, no suelen producir cantidades significativas de estrógenos, aunque en ocasiones pueden elevar transitoriamente sus niveles séricos, determinando un desarrollo mamario transitorio. Un incremento del volumen (> 2 mL), una relación cuerpo/cuello mayor de 1 o la presencia de línea endometrial, son signos sugerentes de niveles elevados de estrógenos circulantes y, por tanto, de inicio puberal.
La ecografía testicular puede ser de utilidad en varones con pubertad precoz periférica, especialmente si existe asimetría testicular, para detectar tumores o restos adrenales, en ocasiones no palpables.
Tratamiento
Pubertad precoz central
El tratamiento de la PP central lo que pretende es, según los casos, revertir, detener o, almenos, enlentecer el desarrollo de los caracteres sexuales secundarios, conservar el potencial de crecimiento y evitar las consecuencias psicosociales y conductuales de una pubertad temprana
El tratamiento de la PP central lo que pretende es, según los casos, revertir, detener o, al menos, enlentecer el desarrollo de los caracteres sexuales secundarios, conservar el potencial de crecimiento y evitar las consecuencias psicosociales y conductuales de una pubertad temprana.
En este caso, el tratamiento busca frenar o suprimir la activación del eje HHG. El tratamiento de elección son los análogos de GnRH (GnRHa) de liberación sostenida (depot). En los casos en los que exista una causa orgánica, deberá hacerse tratamiento etiológico si es posible, aunque este rara vez tiene efecto sobre la evolución de la pubertad.
La administración de GnRHa de liberación sostenida produce, tras una breve estimulación de la liberación de gonadotropinas, una prolongada desensibilización de los receptores hipofisarios de GnRH, con inhibición de la secreción de LH/FSH y, como consecuencia, de la producción y liberación de ES. Los más utilizados son: la triptorelina depot, a la dosis de 80-100 µg/kg, y el acetato de leuprolerina, a la dosis de 150-200 µg/kg, que se administran, ambos, por vía intramuscular cada 25-28 días.
La utilización de los GnRHa en los casos de PPC idiopática con inicio a una edad próxima a los límites considerados normales es controvertida; ya que, los efectos beneficiosos del tratamiento sobre la talla final son, en la mayoría de los pacientes, escasos o nulos. Por ello, no existe un consenso internacional(10) y la indicación de tratamiento se establece de forma individualizada, teniendo en consideración factores psicosociales (repercusión psicológica, familiar, social y conductual) y auxológicos (expectativas de crecimiento, edad ósea, rapidez de progresión de la EO y de los caracteres sexuales secundarios). Tras el inicio de la terapia, la VC puede disminuir considerablemente; en estos casos, algunos estudios han sugerido que la adición de GH al tratamiento con GnRHa podría mejorar la VC y las expectativas de talla final; no obstante, no existen suficientes estudios controlados que demuestren la seguridad y eficacia de esta asociación, para poder establecer una clara indicación.
Otro aspecto controvertido sería el momento más adecuado para suspender el tratamiento con GnRHa. Aunque no existe consenso, en el caso de las niñas, se recomienda que se haga a una edad cronológica de ≈ 11 años y EO de ≈ 12-12,5 años; ya que, por encima de estas edades podría no solo no mejorar, sino incluso empeorar las expectativas de talla adulta. En los varones, la experiencia es muy escasa y suele recomendarse, sin mucha base científica, la suspensión del tratamiento alrededor de los 12 años de edad cronológica y de los 13-13,5 años de EO(11). Pocas semanas o meses después de la suspensión, la pubertad progresa y se recupera la respuesta puberal al estímulo con GnRH. En las niñas, la menarquia se produce, habitualmente, entre 6 y 18 meses después.
Pubertad precoz periférica
Los objetivos del tratamiento de la PPP son similares a los de la PPC; si bien, en este caso, los GnRHa son ineficaces y lo que se utilizan son fármacos que inhiben directamente la producción de ES o su acción sobre los órganos diana.
El tratamiento de la PPP será etiológico en aquellos casos en los que sea posible: quirúrgico (extirpación del tumor ovárico, testicular, suprarrenal o productor de HCG) o médico (tratamiento con hidrocortisona en la hiperplasia suprarrenal congénita, quimioterapia en los suprarrenalomas malignos metastásicos, etc.). En el resto de los casos, el tratamiento será sintomático, con fármacos que, como: ketoconazol, acetato de ciproterona, espironolactona, flutamida, testolactona, letrozole y anastrozole, entre otros, son capaces, por diferentes mecanismos, de reducir o inhibir la producción de ES o de bloquear su acción en los órganos diana(2). En general, estos tratamientos no son demasiado eficaces, rara vez se alcanza con ellos una detención completa en el desarrollo de los caracteres sexuales secundarios y una adecuada talla final y, en algunos casos, sus efectos secundarios son importantes.
PUBERTAD RETRASADA (PR)
Concepto
Se considera la “pubertad retrasada”, cuando no se ha iniciado el desarrollo puberal a una edad 2-2,5 SDS por encima de la edad media de su aparición en la población de referencia(12,13). A efectos prácticos, la ausencia de telarquia en las niñas a una edad de 13 años y la ausencia de incremento del volumen testicular (≥ 4 mL) a los 14 años. Se habla de “pubertad detenida”, cuando la pubertad, iniciada tardíamente o no, no llega a completarse y transcurren más de 4-5 años entre su inicio y el desarrollo gonadal completo en los varones o la menarquia en las mujeres. Por último, se habla de “ausencia de pubertad” o “infantilismo sexual”, cuando la pubertad no llega a iniciarse.
Se considera pubertad retrasada, cuando no se ha iniciado el desarrollo puberal a una edad 2-2,5 SDS por encima de la edad media de su aparición en la población de referencia. A efectos prácticos, la ausencia de telarquia en las niñas a una edad de 13 años y la ausencia de incremento del volumen testicular (≥ 4 mL) a los 14 años
La PR, especialmente en los varones, puede conllevar importantes repercusiones psicosociales que son, con frecuencia, el motivo por el que el paciente acude a la consulta. En una época de extremada sensibilidad y labilidad psicológica, donde la imagen corporal es muy importante para la autoestima del sujeto. La falta de desarrollo puberal y la talla baja, que frecuentemente acompaña al retraso puberal, les hace con frecuencia objeto de burlas y vejaciones por parte de sus compañeros de edad, al tiempo que son relegados de las actividades deportivas y sociales, especialmente de aquellas que conllevan una interrelación con el otro sexo. Como consecuencia, es frecuente que los pacientes desarrollen una mala imagen de sí mismos, baja autoestima, conductas depresivas y tendencia al aislamiento y a una cierta agresividad, alteraciones que, en ocasiones, pueden desembocar en fracaso escolar y en problemas familiares y sociales graves. Por otra parte, la PR puede tener, también, repercusiones físicas, sobre la masa ósea (masa ósea disminuida por el retraso en la aparición de los ES) y auxológicas: alteración de las proporciones corporales (extremidades más largas con relativo acortamiento del segmento superior) y pérdida de talla final. Por motivos desconocidos, un pequeño porcentaje de los pacientes con PR pueden no alcanzar una talla adulta adecuada a su contexto familiar(14).
Etiopatogenia y clínica
Las causas que pueden provocar una PR son múltiples; no obstante, pueden ser fácilmente divididas en cuatro categorías (tabla III):
• Retraso puberal simple. Englobaría aquellos retrasos temporales en el inicio puberal de causa desconocida (idiopáticos) o debidos a factores constitucionales o genéticos (“retraso constitucional del crecimiento y de la pubertad” – RCCP-).
• Retraso puberal secundario a enfermedades crónicas. Serían el resultado de trastornos funcionales en el eje HHG secundarios a múltiples patologías crónicas o endocrinopatías (hipogonadismo hipogonadotropo funcional o transitorio).
• Hipogonadismos hipogonadotropos (HHipo). Serían aquellos pacientes que fracasan en su desarrollo puberal por anomalías en los mecanismos de control hipotálamo-hipofisarios de la pubertad.
• Hipogonadismos hipergonadotropos (HHiper). Serían aquellos pacientes que fracasan en su desarrollo puberal por fallo gonadal primario.
La PR es una situación frecuente, aunque su incidencia real es difícil de establecer. Estadísticamente, debería afectar, al menos, a un 2-3 % de la población. También, debería tener una incidencia similar en ambos sexos, pero, y al contrario de lo que ocurre con la PP, se presenta especialmente en varones (≈ 70 %). En ambos sexos, la causa más frecuente es el simple retraso en su inicio, de etiología familiar o idiopática, que representaría alrededor del 60 % de los casos de PR en varones y del 30 % en mujeres.
Retraso constitucional del crecimiento y de la pubertad
El retraso puberal simple de etiología idiopática o familiar (RCCP) es la causa más frecuente de PR y asociado o no a un componente de talla baja familiar, la causa más frecuente de talla baja en la infancia.
El RCCP es más frecuente en varones, que llegan a consultar por este problema en proporciones de 9:1. Se considera una variante cronológica de la normalidad y, aunque puede presentarse de forma esporádica o idiopática, lo hace habitualmente en un contexto familiar de maduración tardía (60-90 % de los casos). El patrón de herencia es sugerente de una herencia autosómica dominante. Serían niños normales con un patrón madurativo familiar más lento que la media de la población. El cuadro clínico se caracteriza por un hipocrecimiento de inicio postnatal, con un patrón de crecimiento característico, que se acompaña de un retraso en la maduración ósea y en el inicio de la pubertad de 2 a 4 años. Son niños que hasta los 12-18 meses de edad crecen normalmente. A partir de ese momento y hasta los 3-4 años (fenómeno de canalización del crecimiento), experimentan una caída en el ritmo de crecimiento que les lleva a situarse en un carril de crecimiento inferior al que les correspondería para su contexto familiar, con frecuencia próximo o por debajo del percentil 3. Cuando existe un componente familiar importante de talla baja, el hipocrecimiento puede ser muy severo, con tallas por debajo de –3 SDS. A partir de los 3-4 años, los niños crecen a un ritmo normal, aunque habitualmente por debajo del percentil 50 de VC, y tienden a mantener el percentil de talla. Cuando alcanzan la edad en la que habitualmente se produce la pubertad, se observa un nuevo periodo de desaceleración del ritmo de crecimiento, lo que se conoce como “depresión prepuberal de la VC”, que los aleja nuevamente de los percentiles normales hasta que se inicia el estirón puberal. Éste se produce de forma normal aunque tardíamente y el pico de VC suele ser menor; de forma que, es un estirón menos aparente y se ganan menos centímetros que cuando éste se produce a una edad media o temprana, compensándose así el mayor número de años de crecimiento. La talla final se alcanza también tardíamente y suele ser acorde con el contexto familiar, aunque, en alrededor del 15 % de estos pacientes, por causas desconocidas, la talla final se sitúa por debajo de lo esperable para su contexto familiar(15).
Retraso puberal secundario a patología crónica
Prácticamente todas las enfermedades crónicas, si son lo suficientemente importantes en gravedad y duración, repercuten de un modo negativo sobre el crecimiento y la maduración
Prácticamente todas las enfermedades crónicas, si son lo suficientemente importantes en gravedad y duración, repercuten de un modo negativo sobre el crecimiento y la maduración (tabla IV).
Los mecanismos fisiopatológicos que median el retraso puberal en las patologías crónicas son múltiples y varían dependiendo de la enfermedad y de la terapia empleada(16). En la mayoría de los casos, un cierto componente de malnutrición (exceso de pérdidas, disminución de ingesta o aumento de necesidades) suele estar presente, lo que condiciona unas modificaciones hormonales de adaptación que afectan, sobre todo, al eje GH-IGF-1 (resistencia parcial a la acción de la GH, retraso de crecimiento y de la maduración ósea) y al eje HHG (retraso puberal secundario a hipogonadismo hipogonadotropo funcional transitorio). Ambos fenómenos, el hipocrecimiento y la PR se consideran como mecanismos de adaptación a la escasez, absoluta o relativa, de nutrientes. Otros mecanismos fisiopatológicos implicados en la PR dependen de la enfermedad responsable, como son, entre otros: trastornos hidroelectrolíticos, hipoxia crónica, citoquinas inflamatorias, disfunciones hormonales, problemas psicopatológicos y tratamientos crónicos (corticoterapia, transfusiones repetidas, quimioterapia…)
En la mayoría de los casos, el cuadro clínico remeda clínica y hormonalmente al RCCP (hipocrecimiento con retraso de la maduración ósea y del inicio puberal con niveles bajos de gonadotropinas y ES). En otras ocasiones, cuando la enfermedad se manifiesta una vez iniciada la pubertad, puede condicionar una “pubertad detenida” (por ej., anorexia nerviosa), con posterior progresión de los caracteres sexuales secundarios si la enfermedad mejora. Más raramente, algunas patologías crónicas, por la propia enfermedad (hemocromatosis, galactosemia) o por la terapia empleada (quimioterapia, radioterapia, cirugía), pueden a la larga determinar hipogonadismos hipo o hipergonadotropos definitivos con ausencia de desarrollo puberal.
Hipogonadismos hipogonadotropos (HHipo)
Los HHipo son responsables de alrededor del 10 % de los retrasos puberales. Se caracterizan por niveles muy disminuidos o ausentes de las gonadotropinas circulantes, LH y FSH. Pueden ser debidos a defectos congénitos o adquiridos y presentarse aislados o asociados a otras deficiencias hormonales (tabla III).
Los HHipo adquiridos son los más frecuentes y, en su mayoría, debidos a procesos tumorales o infiltrativos que afectan a la región hipotálamo-hipofisaria y que originan deficiencias hipofisarias múltiples(17). La causa más frecuente son los tumores, bien por invasión tumoral directa del área hipotálamo-hipofisaria, o bien como consecuencia de su extirpación quirúrgica o de la radioterapia aplicada para su tratamiento. El más frecuente de estos tumores en la infancia es el craneofaringioma, pero otros tumores, como: germinomas, gliomas o prolactinomas, pueden determinar manifestaciones clínicas similares. La dosis de radioterapia recibida por el hipotálamo o la hipófisis necesaria para producir un HHipo no está claramente establecida, aunque suele ser mayor de 40 Gy. Dosis de 30-55 Gy pueden determinar, inicialmente, una pubertad precoz o adelantada y, más adelante, por el efecto progresivo de la radiación, conducir a un HHipo. Procesos infiltrativos (histiocitosis, sarcoidosis, hemocromatosis), traumatismos craneales, procesos infecciosos o inflamatorios (hipofisitis autoinmune) que afecten al área hipotálamo-hipofisaria son otras posibles causas de HHipo. La hemocromatosis, por acumulo de hierro en hipotálamo-hipófisis puede provocar un HHipo y por acúmulo gonadal un HHiper.
Los hipogonadismos hipogonadotropos se caracterizan por niveles muy disminuidos o ausentes de las gonadotropinas circulantes, LH y FSH
La prevalencia de HHipo congénitos se estima en alrededor de 1:10.000 personas y la mayoría son casos esporádicos, con un predominio en varones 3-5:1(18)Las formas familiares pueden heredarse con carácter autosómico dominante, recesivo o recesivo ligado al X. Los HHipo congénitos pueden presentarse aisladamente, asociados a otras deficiencias hipofisarias o en el contexto de otros cuadros sindrómicos complejos (tabla III).
• HHipo congénitos aislados.- Clásicamente y desde una visión clínica, estas formas de hipogonadismo se han clasificado como “HHipo congénitos con y sin alteraciones del olfato”; no obstante, esta diferenciación puede ser algo artificial, dado que el avance en los estudios genéticos ha puesto de manifiesto, como mutaciones en un mismo gen e incluso en una misma familia puede dar lugar a HHipo con y sin alteraciones del olfato.
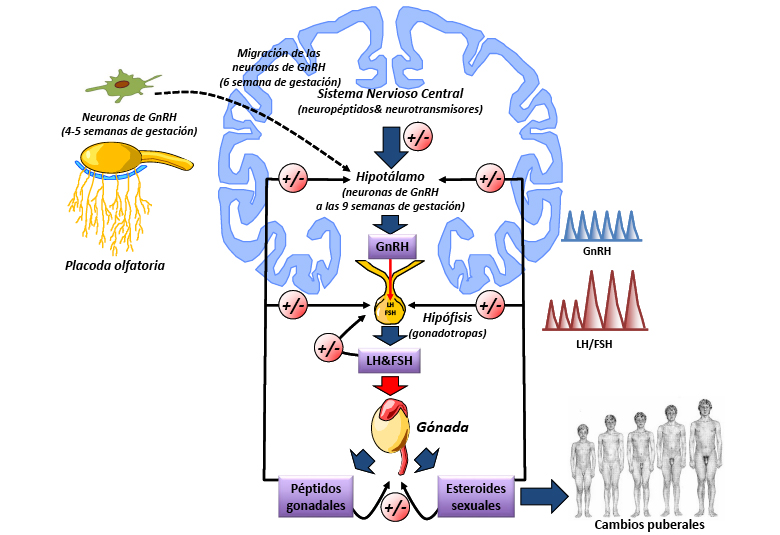
La asociación de HHipo congénitos y alteración del olfato (anosmia o hipoosmia) secundaria a aplasia/hipoplasia de los bulbos olfatorios es lo que se conoce como síndrome de Kallmann (SK). Este síndrome supone alrededor de un 15 % de los HHipo y es cinco veces más frecuente en varones que en mujeres. La asociación de HHipo, por deficiencia de GnRH, y anosmia tiene su explicación en el origen embriológico común de las neuronas productoras de GnRH y de las neuronas olfatorias (Figura 1). Los pacientes con SK pueden mostrar además de los trastornos del olfato, de los que frecuentemente no son conscientes, alteraciones muy variadas, entre ellas: agenesia renal unilateral, defectos atriales septales, ceguera para los colores, hipoacusia neurosensorial, sincinesias de los dedos, lesiones de línea media (labio-paladar hendido, agenesia de uno o más dientes y agenesia del cuerpo calloso), etc. Los casos esporádicos son los más frecuentes, más de dos terceras partes. En los casos familiares, las manifestaciones pueden ser muy variables entre los distintos familiares afectos: anosmia aislada, pubertad tardía, infertilidad, HHipo o pérdida temprana de la menstruación, entre otras. El primer gen responsable del SK, el gen KAL1 (Xp22.31), se descubrió en 1992 y codifica para una proteína, la anosmina, que facilita el crecimiento y la migración neuronal. Desde entonces, se han descrito, al menos, 16 genes diferentes asociados al síndrome (tabla III) que serían responsables en conjunto de menos del 50 % de los SK y con patrones hereditarios diferentes (autosómicos dominante, recesivo y ligado al X).
Las bases genéticas de los HHipo congénitos aislados sin anosmia (HHipo normoósmicos), al igual que ocurre con los SK, son sólo parcialmente conocidas. Algunos de los genes que se han asociado a cuadros de HHipo sin anosmia son: GnRH1 (GnRH), GnRHR (receptor de GnRH), KISS1 (kisspeptina), KISS1R (receptor de las kisspeptinas), LEP (leptina), LEPR (receptor de leptina), PC1 (prohormona convertasa-1), TAC3 (neurokinina B), TAC3R (receptor de TAC3), DAX1 (miembro de la superfamilia de receptores nucleares huérfanos), LHß (subunidad ß de la LH) y FSHß (subunidad ß de la FSH). Las mutaciones en LEP, LEPR y PC1 cursan con obesidad de inicio temprano y las mutaciones en DAX1 se asocian a hipoplasia suprarrenal congénita. Sorprendentemente, alrededor del 10-20 % de estas formas de HHipo aislado congénito, tanto con, como sin anosmia, pueden experimentar cierto grado de reversibilidad espontánea(18).
• HHipo asociados a otras deficiencias hipofisarias.- Son debidas a anomalías congénitas en el desarrollo del SNC (displasia septo-óptica, holoprosencefalia, etc.) de base genética conocida o no, como sería el caso de algunas formas de displasia septo-óptica asociadas a mutaciones en HEX1, o a mutaciones en factores de transcripción implicados en el desarrollo de las diferentes líneas células hipofisarias: LHX3, LHX4, PROP1 y POU1F1 (Pit1). El gen más frecuentemente afectado es PROP1, cuyas mutaciones determinan una deficiencia combinada de: GH, Prl, LH, FSH, TSH y, frecuentemente, pero de forma más tardía, también de ACTH.
• HHipo asociados a cuadros sindrómicos.- Determinados síndromes pueden asociar cuadros clínicos de HHipo, como es el caso de los síndromes de: Prader-Willi, CHARGE (cuando es por mutaciones en CDH7, se puede asociar anosmia), Laurence-Moon, Bardet-Biedl, 4H, entre otros.
Las manifestaciones clínicas de los HHipo son variables dependiendo de su etiopatogenia y momento de aparición (congénitos, infancia, pubertad o edad adulta), como sería el caso de: trastornos del olfato, rasgos sindrómicos, anomalías congénitas asociadas, síntomas debidos a otras deficiencias hipofisarias (GH, ADH, ACTH…), etc. En general, las formas congénitas, dado que la diferenciación sexual está controlada por la gonadotropina coriónica placentaria (HCG) y no por las gonadotropinas fetales, no presentan ambigüedad genital, aunque sí condicionan con frecuencia criptorquidia bilateral con micropene o una hipoplasia de labios menores, debido a su acción en la fase final de la gestación. Durante el periodo prepuberal, el crecimiento y la maduración ósea suelen ser normales, pero al llegar a la edad puberal, la ausencia de ES determina que no se desarrollen los caracteres sexuales secundarios (puede aparecer vello pubiano escaso por acción de los andrógenos suprarrenales) y se retrase el cierre de los cartílagos de crecimiento, lo que favorece el desarrollo de proporciones corporales eunucoides (aumento de las extremidades con incremento de la braza, > 5 cm que la talla, y disminución del cociente segmento superior/segmento inferior). Algunos pacientes pueden iniciar la pubertad y no completarla (pubertad detenida) o completarla y desarrollar el hipogonadismo, posteriormente, en la edad adulta, manifestándose en forma de infertilidad y disfunción sexual.
Hipogonadismos hipergonadotropos (HHiper)
Los hipogonadismos hipergonadotropos son debidos a fallo gonadal primario y se caracterizan por niveles séricos elevados de gonadotropinas y disminuídos de ES
Este tipo de hipogonadismos son debidos a fallo gonadal primario y se caracterizan por niveles séricos elevados de gonadotropinas y disminuídos de ES. Pueden ser congénitos o adquiridos (Tabla III). La incapacidad de la gónada para producir adecuadas cantidades de ES e inhibina determina la falta de retroinhibición de estos sobre el hipotálamo y la hipófisis, incrementándose la secreción pulsátil de GnRH y, por tanto de LH y FSH.
Las dos causas más frecuente de HHiper congénito son dos cromosomopatías congénitas: el síndrome de Klinefelter y el síndrome de Turner.
• El síndrome de Klinefelter o síndrome de disgenesia de los túbulos seminíferos (47, XXY y sus variantes) es la causa más frecuente de hipogonadismo en el varón (1:500-1.000 niños nacidos vivos). La función de los túbulos seminíferos y de las células de Leydig está alterada y la espermatogénesis ausente. Dependiendo de los niveles de testosterona, la pubertad puede desarrollarse normalmente, retrasarse o empezar a una edad normal, aunque sin una adecuada progresión. Las manifestaciones clínicas son variables, pero la talla suele ser alta y en la pubertad y edad adulta los testes son pequeños y duros y la ginecomastia es frecuente. Otras anomalías asociadas incluirían: retraso mental variable, dificultades en el lenguaje, problemas de conducta, incremento en la incidencia de determinados tumores (cáncer de mama y tumores de células germinales de localización mediastínica, retroperitoneal y pineal) y alteraciones tiroideas, entre otras.
• El síndrome de Turner (45, X0 y sus variantes) es la causa más frecuente de HHiper en la mujer (1:2.500-3.000 niñas nacidas vivas). Sus manifestaciones clínicas resultan de la ausencia de genes que escapan a la inactivación del X. Estas niñas pueden presentar diferentes alteraciones y anomalías, incluyendo: hipocrecimiento, fallo gonadal, rasgos sindrómicos (pterigium colli, linfedema, tórax en coraza, hipoplasia areolar, cubitus valgo, alteraciones ungueales, acortamiento de metacarpianos, implantación baja del cabello y de las orejas, boca de pez, nevus múltiples, etc.), cardiopatía, malformaciones del sistema urinario, etc. Los síntomas más constantes son el hipocrecimiento (95 %)y el fallo gonadal (90 %). Pese a ello, un 30% son capaces de iniciar espontáneamente la pubertad, aunque sólo un 2-5% llegan a completarla y a tener reglas espontáneas (menopausia precoz con amenorrea secundaria).
• Las causas adquiridas de HHiper son relativamente infrecuentes: torsión gonadal bilateral (testicular u ovárica), castración quirúrgica (tumores), traumatismos severos en el escroto y testículos, orquitis bilaterales (por ej. parotiditis) y, en el caso de las mujeres, galactosemia o fracaso ovárico precoz de etiología idiopática o autoinmune. El tratamiento del cáncer, debido a la quimioterapia y radioterapia, con frecuencia aplicadas conjuntamente, es una causa creciente de HHiper adquirido.
Evaluación diagnóstica
Las causas que pueden provocar un retraso puberal son múltiples. Una valoración básica inicial incluiría: una historia personal y familiar, una exploración física completa y una serie de pruebas complementarias que pueden variar en función de los hallazgos de la historia y exploración(19).
Anamnesis detallada
Una historia familiar de PR está presente en la gran mayoría de los casos de RCCP, pero también puede formar parte de la historia familiar en pacientes con HHipo. Un interrogatorio cuidadoso puede poner de manifiesto la presencia de síntomas sugerentes de patologías concretas (anosmia, infertilidad, galactorrea, hipotiroidismo, etc.) o de patologías crónicas inadvertidas, así como un exceso de ejercicio o un trastorno de la conducta alimentaria. Los antecedentes de criptorquidia, quimioterapia, radioterapia gonadal o craneal y la administración crónica o prolongada de medicamentos que puedan interferir en la función gonadal debe ser, también, recogida. La reconstrucción de la gráfica de crecimiento y peso puede ser de utilidad, junto con los datos auxológicos parentales. El hipocrecimiento es un hallazgo habitual en: RCCP, malnutrición, patología crónica o Turner; mientras que, en la mayoría de las formas de HHipo la talla suele ser normal o incluso alta. Un bajo peso para la talla puede indicar trastornos nutricionales o enfermedad crónica inadvertida; mientras que, hipotiroidismo, exceso de glucocorticoides, deficiencia de GH o determinados síndromes (Prader Willi, Turner, etc.), suelen tener un claro o moderado incremento del peso para la talla.
Exploración física
Debe ser completa, analizando especialmente: talla, peso, signos de malnutrición o patología crónica, estigmas sindrómicos (Turner, Klinefelter…) y signos neurológicos sugerentes de patología intracraneal (fondo de ojo, campimetría y estudio del olfato pueden ser necesarios). Debe realizarse una cuidadosa valoración del estadio de desarrollo puberal; ya que, signos incipientes de desarrollo puberal pueden pasar inadvertidos para los pacientes o alteraciones en la secuencia normal de la pubertad pueden sugerir patología. En las niñas con desarrollo puberal normal, pero sin menarquia, deben descartarse causas anatómicas de amenorrea (himen imperforado, septum transverso vaginal o disgenesia mülleriana -síndrome de Rokitansky-), mediante una adecuada exploración ginecológica y ecográfica.
Edad ósea (EO)
El RCCP, la patología crónica, las endocrinopatías y los hipogonadismos presentan, habitualmente, un retraso de EO de 1 a 4 años. Una talla normal-baja, con ralentización reciente y EO inferior a 11 años en una niña y a 13 años en un varón es muy sugerente de retraso puberal simple; por el contrario, la ausencia de signos puberales a una EO de más de 11 años en las niñas y de más de 13 años en los niños es muy sugerente de hipogonadismo.
Otras pruebas complementarias
Su realización dependerá de la historia, exploración y EO.
• Analítica general. En pacientes en los que la historia clínica o la exploración física sugieran la presencia de patología crónica subyacente, debe realizarse una evaluación individual orientada a la sospecha clínica. Esta puede incluir: hemograma y bioquímica básica, marcadores de enfermedad celíaca, anticuerpos antitiroideos, TSH, T4 libre, prolactina y marcadores de deficiencia de GH (IGF-I, IGFBP-3). La sospecha de una deficiencia de GH puede obligar a realizar test de GH precedidos de la administración de ES (primación) para diferenciar una deficiencia real de GH de una deficiencia transitoria asociada a RCCP.
• Cariotipo. Su realización estaría indicada ante la presencia de estigmas sindrómicos o en el caso de: gonadotropinas elevadas, niñas con talla baja de etiología incierta o varones con testes pequeños e inadecuados para el grado de desarrollo puberal.
• RM craneal. Podría poner de manifiesto patología orgánica intracraneal, en área hipotálamo-hipofisaria, o anomalías de la vía olfatoria (agenesia de bulbos o nervios olfatorios).
• Ecografías pélvico-abdominales. Podrían poner de manifiesto malformaciones congénitas asociadas (renales en el SK) o anomalías vaginouterinas (agenesia o malformaciones uterinas, septum vaginal…), tamaño y morfología del útero y ovarios, localización de testes criptorquídicos…
• Estudios hormonales. Un aspecto fundamental de la evaluación del retraso puberal, pero especialmente compleja, es la valoración del eje HHG. Los niveles séricos de testosterona y estradiol son de escasa utilidad en las fases iniciales de la pubertad; ya que, sus niveles séricos se sitúan, con frecuencia, por debajo del límite de detección de la mayoría de los inmunoanálisis. A partir de los 10-11 años de EO, a veces incluso antes, es frecuente observar en los HHiper niveles séricos elevados de LH y FSH basales o tras estímulo con GnRH. El diagnóstico de los HHipo completos también es sencillo cuando la EO supera, al menos en un año, la EO en la que habitualmente la pubertad se inicia. Se observan en este caso, niveles séricos disminuidos de LH y FSH tras estímulo con GnRH. El principal problema de diagnóstico diferencial se plantea entre el RCCP y el HHipo (sobre todo si es parcial, aislado e idiopático) cuando la EO del paciente está retrasada por debajo de las edades en que normalmente se inicia la pubertad. En estos casos, existe un considerable solapamiento entre la pobre respuesta de los pacientes con RCCP y la observada en pacientes con HHipo. Inhibina B y hormona antimülleriana, marcadores de la célula de Sertoli, pueden ser en ocasiones de utilidad para diferenciar HHipo y RCCP; ya que, la presencia de niveles séricos de inhibina B y hormona antimülleriana muy disminuidos o ausentes es más característico de los HHipo que de los RCCP. No obstante, en muchos casos, solo el tiempo y la evolución espontánea de la pubertad permitirán excluir o confirmar, definitivamente, el hipogonadismo(15).
• Estudios moleculares. Los pacientes con hipogonadismo y cariotipo normal, así como los hipogonadismos sindrómicos son candidatos para estudios moleculares más amplios. Estos deberían estar orientados, en función del diagnóstico y los hallazgos clínicos y hormonales, hacia genes concretos. En el caso de que las características clínicas del paciente no orienten hacia un diagnóstico concreto, se puede considerar, en colaboración con el genetista clínico, la posibilidad de realizar otros estudios genéticos más amplios: estudios de MLPA (Multiplex Ligation-dependent Probe Amplification), GWAS (Genoma-Wide Association Studies) o estudios de secuenciación de alto rendimiento (NGS: Next-Generation Sequencing). Este último tipo de estudios permite la secuenciación del genoma completo, de capturas selectivas (paneles de genes implicados en determinadas patologías), de regiones concretas o de todas las regiones codificantes y reguladoras conocidas de todo el genoma (exoma).
Los pacientes con hipogonadismo y cariotipo normal, así como los hipogonadismos sindrómicos son candidatos para estudios moleculares más amplios
Tratamiento
Retraso constitucional del crecimiento y de la pubertad
El RCCP se considera una variante de la normalidad; por consiguiente, en la gran mayoría de los casos, una clara explicación al paciente y a los padres, junto con un adecuado control y apoyo psicólogico, son suficientes. Sólo aquellos casos en los que el retraso sea más severo y existan graves repercusiones psicológicas y sociales (depresión, baja autoestima, fracaso escolar, etc.) serán susceptibles de tratamiento.
En los varones, suele administrarse testosterona, a dosis baja, en forma de preparados depot de ésteres de testosterona (enantato o cipionato), en una inyección intramuscular mensual de 50-100 mg, y a partir de los 12-12,5 años de EO o de los 14 de edad cronológica(20). Por debajo de esa edad, rara vez es necesario desde el punto de vista psicológico y el riesgo de acelerar la EO y comprometer la talla definitiva es mayor. La testosterona acelera la VC, el desarrollo de los caracteres sexuales secundarios y favorece el desarrollo espontáneo de la pubertad. Se recomienda realizar ciclos de tratamiento de 3-6 meses, alternando con periodos similares de observación durante los cuales se vigila la progresión espontánea de la pubertad. Si tras 2-3 ciclos, la pubertad no se ha iniciado (aumento del volumen testicular), lo más probable es que nos encontremos ante un hipogonadismo hipogonadotropo.
En las niñas, la incidencia de RCCP es muy inferior a la de los varones y la experiencia menor. Se recomienda que el tratamiento no se inicie antes de los 13 años de edad cronológica y de los 11-12 años de EO y que se utilicen estrógenos (estradiol o etinil-estradiol) a dosis muy bajas, al objeto de no acelerar en exceso la maduración ósea y comprometer la talla final.
En pacientes con RCCP con malas expectativas de talla adulta se ha sugerido la posibilidad de emplear otro tipo de tratamientos, como sería el caso de la GH; sin embargo, al menos con las pautas utilizadas, la GH no parece ser capaz de mejorar significativamente la talla final en estos pacientes. Otros estudios más recientes sugerían que los inhibidores de la aromatasa de 3ª generación (letrozol y anastrozol) asociados a andrógenos podrían acelerar la VC, enlentecer la progresión de la maduración ósea y mejorar las expectativas de talla final y, todo ello, sin efectos secundarios reseñables; no obstante, los resultados disponibles a talla final, aunque todavía insuficientes para poder establecer conclusiones definitivas, no sostienen estos potenciales beneficios y sí posibles efectos secundarios, entre ellos: marcada elevación de los niveles séricos de testosterona, de repercusión incierta, desarrollo de anomalías vertebrales, disminución del colesterol HDL y aumento del hematocrito(21).
Retraso puberal secundario a patología crónica
El tratamiento y la prevención del retraso puberal en pacientes con patologías crónicas se basa en el tratamiento óptimo y precoz de la enfermedad de base, junto con una adecuada nutricion (aporte suficiente de macro y micronutrientes).
Las pautas para inducir y mantener el desarrollo puberal no difieren, en general, de las empleadas en el RCCP o en el hipogonadismo.
Hipogonadismos
En los hipogonadismos es necesario inducir o completar el desarrollo de los caracteres sexuales secundarios y posteriormente, establecer una pauta crónica de reemplazamiento de ES
En los hipogonadismos es necesario inducir o completar el desarrollo de los caracteres sexuales secundarios y posteriormente, establecer una pauta crónica de reemplazamiento de ES.
Disponemos de tres opciones terapéuticas para inducir la pubertad: administración pulsátil de GnRH, administración de gonadotropinas y administración de ES. La elección dependerá del diagnóstico (en los HHiper, la única opción son los ES), de los objetivos terapéuticos (incremento de volumen testicular, fertilidad ), de la disponibilidad (la terapia con GnRH está limitada a muy pocos centros en Europa) y de las preferencias del paciente.
Durante la infancia, en los varones con HHipo puede ser necesario tratamiento para resolver la criptorquidia y mejorar el micropene. La criptorquidia, sobre todo cuando es bilateral, puede tener consecuencias negativas para la fertilidad futura, por ello se recomienda su corrección quirúrgica temprana, preferiblemente entre los 6-12 meses de vida(18). En cuanto al micropene, puede ser tratado con pequeñas dosis de testosterona depot (25 mg/mes), 1-3 dosis, preferiblemente durante los primeros 6-12 meses de vida o con gonadotropinas (HCG y FSH). Estudios recientes sugieren que el tratamiento con gonadotropinas en este periodo de la vida podría tener, además, efectos beneficiosos sobre el desarrollo testicular y la futura fertilidad del paciente, aunque estos resultados son todavía muy preliminares para establecer conclusiones definitivas.
En la adolescencia y edad adulta, los objetivos del tratamiento son: conseguir un desarrollo puberal completo, con una secuencia y ritmo normal de aparición de los caracteres sexuales secundarios, lograr la mejor talla adulta posible, evitar la osteoporosis y las complicaciones cardiovasculares y psicosociales, y, en aquellos casos en que sea factible, adquirir y optimizar la capacidad reproductiva(22,23). Una propuesta aceptable sería, remedando el desarrollo puberal normal, inducirlo alrededor de los 11 años de EO en las niñas y de los 12 años en los varones e incrementar lentamente los niveles séricos de ES para conseguir el desarrollo puberal completo en un periodo de 3-4 años. Cuando la talla final está comprometida (deficiencia de GH, Turner), puede ser necesario retrasar la inducción de la pubertad, al objeto de lograr unas mejores expectativas de talla adulta.
En varones, la forma más sencilla de inducir el desarrollo puberal es la administración de testosterona, que puede ser por vía oral (no disponible en España), parenteral o transdérmica. En la práctica, suelen utilizarse preparados depot de testosterona de acción prolongada (enantato o cipionato) por vía intramuscular. La dosis inicial será de 25-50 mg cada 4 semanas y se incrementará en 50 mg, cada 6-12 meses, para, a lo largo de un periodo de 3-4 años, alcanzar la dosis de sustitución de un adulto, que oscilaría entre 200-250 mg cada 10-14 días. Un inconveniente de esta terapia es que el volumen testicular no aumenta, tampoco, ni se induce la fertilidad. En los niños con HHipo en los que se desee incrementar el volumen testicular, la terapia intramuscular o subcutánea con gonadotropinas o la administración pulsátil, mediante bomba, de bolos de GnRH, por vía subcutánea, puede ser una alternativa. Una vez completado el desarrollo puberal, la terapia de mantenimiento en los varones se realiza, habitualmente, con testosterona. Aunque en este caso, también puede administrarse por diferentes vías, las más utilizadas son: la intramuscular (200-250 mg de enantato de testosterona cada 10-14 días o 1000 mg de undecanoato detestosterona c/2-3 meses) y la transdérmica (parches de testosterona); ya que, los preparados orales de testosterona (undecanoato de testosterona) no está comercializados en nuestro país(21). La fertilidad, en el adulto, requiere de la administración gonadotropinas o de bolos de GnRH, por vía subcutánea, durante periodos prolongados para inducir la espermiogénesis.
En las niñas, la inducción de la pubertad se realizará con estrógenos y con dosis iniciales muy bajas; ya que, los estrógenos son un potente inductor de la fusión epifisaria. Los regímenes más habitualmente empleados incluyen la administración oral o transdérmica de estradiol. La vía transdérmica, con parches de composición matricial es la más recomendada, ya que tiene la ventaja sobre la vía oral, de una mayor biodisponibilidad, mejor tolerancia gastrointestinal y menor toxicidad hepato-biliar al evitar el paso inicial por el hígado. Estos parches matriciales, dependiendo del preparado, liberan 25, 50, 75 o 100 µg/día de estradiol, pero permiten su fragmentación y la administración de dosis más bajas y progresivamente crecientes de estradiol. La dosis inicial sería de 0,05-0,1 µg/kg, habitualmente 1/8 de parche de 25 µg que, de forma ideal, se aplicaría durante los primeros 3-4 meses solo por la noche (se pondría al acostarse y se retiraría por la mañana); posteriormente, el 1/8 de parche se mantendría durante todo el día (se cambian 2 veces/ semana) y las dosis se irían incrementando cada aproximadamente 6-12 meses, durante un periodo no inferior a 2-3 años, hasta alcanzar la dosis diaria de sustitución estrogénica de una mujer adulta, que correspondería habitualmente a unos 50 µg/día de estradiol(22)..Una vez alcanzado un adecuado desarrollo mamario (T4-5) y uterino (útero en la ecografía de >35-40 mm, con línea endometrial visible) o bien si produce un sangrado menstrual o pequeños manchados, debe añadirse un progestágeno cíclico, preferentemente micronizado para favorecer su absorción (200 mg/día, antes de acostarse, del 10-21 día de ciclo, en forma de cápsulas orales u óvulos vaginales) para la protección uterina y establecer ciclos menstruales regulares mensuales. Una vez completado el desarrollo puberal, es necesario establecer una terapia sustitutiva a largo plazo. Se podría seguir con la asociación de estrógeno transdérmico y progestágeno oral-vaginal, pero, en la mayoría de los casos, suele administrase una combinación continua o cíclica de estrógenos-progestágenos, habitualmente por vía oral o transdérmica. Una gran variedad de preparados combinados de estrógenos-progestágenos están disponibles en el mercado en forma de píldoras orales anticonceptivas y pueden ser empleados. Deben elegirse, si están disponibles, aquellos que contengan estrógenos naturales y, si no es posible, los de menor contenido estrogénico (15-20 µg de etinil estradiol). La opción a la vía oral es la administración transdérmica continua de estrógenos-progestágenos que, habitualmente, aportan una dosis diaria de 50 µg/día de estradiol. Los parches se cambian cada 3,5 días (dos por semana), durante 3 semanas, transcurridas las cuales se suspende una semana el tratamiento, durante la cual se produce la regla. La inducción de fertilidad, requiere, como en el caso de los varones, de la administración prolongada de gonadotropinas o GnRH.
Tablas y figuras
Tabla I. Etiopatogenia de la pubertad precoz (PP)
|
PP central |
PP periférica |
|
Idiopática • Esporádica • Familiar • Tras adopción internacional Genética: • Mutaciones activadoras en KISS1 • Mutaciones activadoras en KISS1R (GPR54) • Mutaciones inactivantes en MKRN3 (de origen paterno) Secundaria a alteraciones del SNC: • Tumores: — Hamartoma hipotalámico — Craneofaringioma — Otros: astrocitoma, glioma, ependimoma, pinealoma, neuroblastoma, adenomas secretores de gonadotropinas*, etc. • Anomalías congénitas: — Hidrocefalia — Mielomeningocele — Defectos del desarrollo del cerebro medio • Lesiones quísticas: — Quiste aracnoideo, glial o pineal — Quiste hidatídico • Infecciones: — Meningitis, encefalitis y abscesos • Irradiación craneal • Lesiones vasculares • Lesiones del SNC de otro tipo Asociada a determinados cuadros sindrómicos: • Neurofibromatosis tipo I • S. de Russel-Silver • S. de Beuren-Williams • S. de Cohen • Disomía uniparental materna del PP central tras exposición a ES (PP mixta) |
Función gonadal autónoma — S. de McCune-Albright (gen Gsα) — PP familiar del varón o testotoxicosis (LHR) — Quistes ováricos Tumores gonadales: — Ovario: — Células de la granulosa — Células de la teca — Celularidad mixta — Testículo: — Células de Leydig — Células de Sertoli (asociación a — Otros: restos adrenales, etc. Exposición o ingestión de ES exógenos Tumores secretores de HCG — Hepatoblastoma — Pinealoma — Germinoma — Coriocarcinoma — Teratoma — Otros Patología suprarrenal: — Hiperplasia suprarrenal congénita — Corticosuprarrenaloma (adenoma o carcinoma) Hipotiroidismo primario severo Resistencia generalizada a los glucocorticoides |
|
SNC: sistema nervioso central. S: síndrome. ES: esteroides sexuales. HCG: gonadotropina coriónica. S: síndrome; Gsα: gen de la subunidad alfa de las proteínas G de membrana; LHR: gen del receptor para la LH; KISS1: gen de las kisspeptinas; KISS1R: gen del receptor de la kisspeptinas. * Aunque se ha sugerido esta posibilidad, no hay evidencia sólida de que los adenomas hipofisarios secretores de gonadotropinas puedan ocasionar un PP. |
|
Tabla II. Criterios para ayudar a diferenciar entre una verdadera pubertad precoz (PP) central y telarquia prematura aislada (variante normal de la pubertad) en niñas*
|
PP central verdadera |
Telarquia prematura aislada |
|
|
Criterios clínicos |
||
|
• Progresión a través de |
Progresión de un estadio al |
Estabilización o regresión de los signos puberales |
|
• Velocidad de crecimiento |
Acelerada (≥ 6 cm/año) |
Habitualmente normal para la edad |
|
• Edad ósea |
Avanzada, habitualmente |
Normal o avanzada < 1 año |
|
• Predicción de talla adulta |
Inferior a talla diana o se reduce en las predicciones de talla seriadas |
Dentro del rango de la talla diana |
|
Criterios ecográficos |
||
|
• Ecografía pélvica |
Volumen uterino > 2 mL, longitud > 34 mm, forma de pera o presencia de línea endometrial |
Volumen uterino ≤ 2 mL, longitud ≤ 34 mm, forma tubular prepuberal |
|
Criterios hormonales |
||
|
• Estradiol |
Niveles séricos aumentados con el avance del desarrollo puberal. |
Niveles séricos indetectable o próximos al límite de detección |
|
• Test de LHRH |
Patrón puberal |
Patrón prepuberal o intermedio |
Tabla III. Etiopatogenia de la pubertad retrasada
|
Retraso puberal simple (RCCP) |
Hipogonadismos hipergonadotropos: |
|
• Familiar • Esporádico/idiopático |
• Congénitos: • Varones: – S. de Klinefelter (XXY) – Disgenesia gonadal (XO/XY) – Defectos de la biosíntesis/acción de T1 – Errores innatos en la síntesis de T1 – Déficit de 5 alfa-reductasa – SIPA parcial – Hipoplasia/agenesia de las células de Leydig – Mutaciones genes de los receptores de LH o FSH – Anorquia (S. de los testículos evanescentes) – S. polimalformativos (S. Noonan, distrofia miotónica, etc.) • Mujeres: – S. Turner (XO) – Disgenesia gonadal (XO/XY o XX) – SIPA completa (S. Morris) – S. polimalformativos • Adquiridos: • Varones: – Orquitis bilateral (parotiditis, etc.) • Mujeres: – Fallo ovárico precoz autoinmune – Galactosemia • Ambos: – Hemocromatosis – Castración quirúrgica o traumática – Torsión gonadal bilateral – Radioterapia/Quimioterapia |
|
Retraso puberal 2rio a patologías crónicas (tabla IV) |
|
|
Hipogonadismos hipogonadotropos (HH): |
|
|
• Congénitos aislados: • HH con anosmia (S. Maestre de San Juan-Kallmann): – Mutaciones en: KAL1, FGFR1, PROKR2, PROK2, CHD7, FGF8, NELF, WDR11, HS6ST1, SEMA3A, SPRY4, IL17RD, DUSP6, FGF17, FLRT3 y FEZF1 (muchos de ellos puedan ocasionar también HH sin anosmía, incluso en las mismas familias) – Idiopático • HH sin anosmia: – GnRH1 (GnRH), – GnRHR (receptor de GnRH) – KISS1 y KISS1R (kisspeptinas y su receptor) – LEP y LEPR (leptina y su receptor) – PC1 (prohormona convertasa-1), – TAC3 y TAC3R (neurokinina B y receptor) – DAX1 (HH asociado a hipoplasia suprarrenal) – LHß y FSHß (subunidad ß de LH y FSH) – Idiopático • Congénitos asociados a otras deficiencias hipofisarias: • Mutaciones en LHX3, LHX4, PROP1 y POU1F1 – HH asociado a anomalías congénitas en el SNC – Esporádicas – Asociadas a cromosomopatías o defectos génicos (HESX-1, ZIC-2) – HH asociado a cuadros sindrómicos (Prader Willi, CHARGE, Bardet-Beidl, etc.) • Adquiridos por lesión hipofisaria, habitualmente asociados a otras deficiencias hipofisarias: – Tumores selares o extraselares (craneofaringiomas, germinomas, gliomas, etc.) – Histiocitosis/Sarcoidosis – Hemocromatosis – Hipofisitis autoinmune – Apoplejía hipofisaria – Lesiones postinfecciosas (meningitis, tuberculosis, etc.) – Lesiones postquirúrgicas o postraumáticas – Lesiones postradiación |
|
|
T1: testosterona. SIPA: síndrome de insensibilidad periférica a los andrógenos. FSHR: receptor de FSH. LHR: receptor de LH. LHß: subunidad ß de la LH. FSHß: subunidad ß de la FSH. S: síndrome. SNC: sistema nervioso central. |
|
Tabla IV. Principales patologías crónicas responsables de retraso puberal
|
• Malnutrición • Infecciones recurrentes/Infestaciones crónicas • Inmunodeficiencias: – Congénitas – SIDA • Enfermedades gastrointestinales: – Malabsorción: * Enfermedad celíaca * Infestación por Giardia Lamblia * Fibrosis quística – Enfermedad inflamatoria intestinal – Hepatopatías crónicas • Enfermedades renales: – Nefropatías glomerulares – Tubulopatías congénitas – Nefropatías intersticiales – Síndrome nefrótico – Insuficiencia renal crónica • Enfermedades respiratorias: – Asma crónico – Fibrosis quística |
• Enfermedades hematológicas: – Anemias crónicas – Histiocitosis – Hemocromatosis • Endocrinopatías: – Deficiencia de hormona de crecimiento – Hipotiroidismo/hipertiroidismo – Diabetes mellitus tipo 1 mal controlada – Hipercortisolismo – Hiperprolactinemia • Trastornos de la conducta alimentaria: – Anorexia nerviosa – Bulimia nerviosa • Ejercicio excesivo (amenorrea atlética) • Patología oncológica • Miscelánea: – Enfermedades inflamatorias del tejido conectivo – Enfermedades neurológicas – Estrés psicológico – Enfermedad de Gaucher – Cardiopatías crónicas – Consumo de marihuana |
Figura I. Eje hipotálamo-hipófiso-gonadal (HHG)
Algoritmo
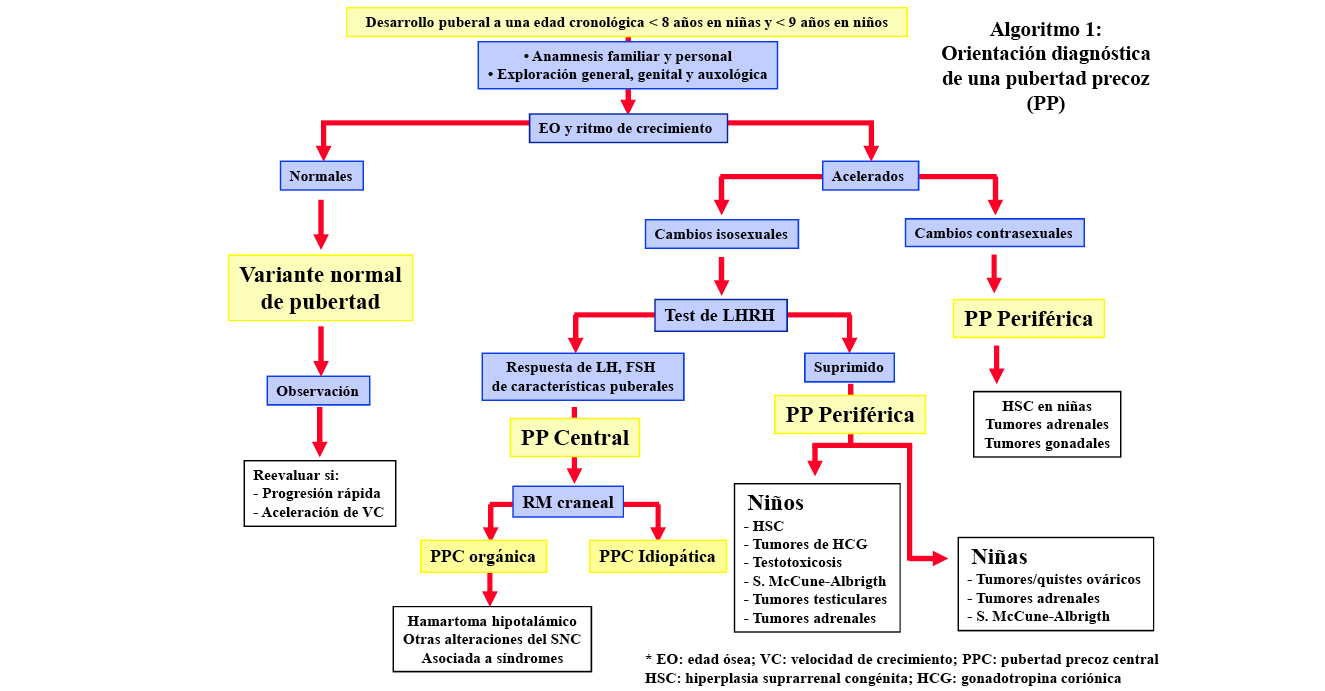
Bibliografía
1. Carel JC, Léger J. Precocious puberty. N Engl J Med. 2008; 358: 2366-77.
2. M, Chaussain Carel JC, LahlouN, Roger JL. Precocious puberty and statural growth. Human Reprod changes after migration. Endocr Rev 2003; 24: 668-93
3. Johansson T, Ritzzen EM. Very long-term follow-up of girls with early and late menarche. Endocr Dev 2005; 8: 126-36.
4. Latronico AC, Brito VN, Carel JC. Causes, diagnosis, and treatment of central precocious puberty. Lancet Diabetes Endocrinol. 2016; 4(3):265-74.
5. Shin YL. An update on the genetic causes of central precocious puberty. Ann Pediatr Endocrinol Metab. 2016;21(2):66-9.
6. Stephen MD, Zage PE, Waguespack SG. Gonadotropin-dependent precocious puberty: neoplastic causes and endocrine considerations. Int J Pediat Endocrinol. 2011; 2011: 184502. doi:10.1155/2011/184502.
7. Barrio R, Carcavilla A, Martín M. Pubertad Precoz y Retrasada. Inf Ter Sist Nac Salud. 2006; 30: 95-107.
8. Dumitrescu CE, Collins MT. McCune-Albright syndrome. Orphanet J Rare Dis. 2008; 3: 12.
9. Soriano-Guillén L, Corripio R, Labarta JI, et al. Central precocious puberty in children living in Spain: incidence, prevalence, and influence of adoption and immigration. J Clin Endocrinol Metab. 2010; 95: 4305-13
10. Pienkowski C, Tauber M. Gonadotropin-Releasing Hormone Agonist Treatment in Sexual Precocity. Endocr Dev. 2016;29:214-29.
11. Guaraldi F, Beccuti G, Gori D1, Ghizzoni L. MANAGEMENT OF ENDOCRINE DISEASE: Long-term outcomes of the treatment of central precocious puberty. Eur J Endocrinol. 2016;174(3):R79-87.
12. Dunkel L, Quinton R. Transition in endocrinology: induction of puberty. Eur J Endocrinol. 2014;170(6):R229-39.
13. Villanueva C, Argente J. Pathology or normal variant: what constitutes a delay in puberty? Horm Res Paediatr. 2014; 82(4): 213-21.
14. Wei C, Crowne EC. Recent advances in the understanding and management of delayed puberty. Arch Dis Child. 2016;101(5):481-8.
15. Harrington J, Palmert MR. Clinical review: Distinguishing constitutional delay of growth and puberty from isolated hypogonadotropic hypogonadism: critical appraisal of available diagnostic tests. J Clin Endocrinol Metab. 2012;97(9):3056-67.
16. Pozo J, Argente J. Delayed puberty in chronic illness. Baillieres Best Pract Res Clin Endocrinol Metab. 2002; 16: 73-90.
17. Nakamoto JM, Franklin SL, Geffner ME. Puberty. En: Kappy MS, Allen DB, Geffner ME, ed. Pediatric Practice Endocrinology. New York: Mc Graw Hill Medical; 2010. p. 257-98.
18. Boehm U, Bouloux PM, Dattani MT, de Roux N, Dodé C, Dunkel L, et al. Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism-pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2015 (artículo publicado electrónicamente previo a su publicación en papel). doi:10.1038/nrendo.2015.112
19. Abitbol L, Zborovski S, Palmert MR. Evaluation of delayed puberty: what diagnostic tests should be performed in the seemingly otherwise well adolescent? Arch Dis Child. 2016;101(8):767-71.
20. Pozo J, Argente J. Ascertainment and treatment of delayed puberty. Horm Res. 2003; 60 (suppl 3): 35-48.
21. Shams K, Cameo T, Fennoy I, et al. Outcome analysis of aromatase inhibitor therapy to increase adult height in males with predicted short adult stature and/or rapid pubertal progress: a retrospective chart review. J Pediatr Endocrinol Metab. 2014; 27: 725-30.
22. Howard S, Dunkel L. Sex Steroid and Gonadotropin Treatment in Male Delayed Puberty. Endocr Dev. 2016;29:185-97.
23. Rastrelli G, Vignozzi L, Maggi M. Different Medications for Hypogonadotropic Hypogonadism. Endocr Dev. 2016;30:60-78.

